

In quantum mechanics, an atomic orbital (/ˈɔːrbɪtəl/) is a function describing the location and wave-like behavior of an electron in an atom.[1] This function describes the electron's charge distribution around the atom's nucleus, and can be used to calculate the probability of finding an electron in a specific region around the nucleus.[2]
Each orbital in an atom is characterized by a set of values of the three quantum numbers n, ℓ, and mℓ, which respectively correspond to the electron's energy, its orbital angular momentum, and its orbital angular momentum projected along a chosen axis (magnetic quantum number). The orbitals with a well-defined magnetic quantum number are generally complex-valued. Real-valued orbitals can be formed as linear combinations of mℓ and −mℓ orbitals, and are often labeled using the associated harmonic polynomials (e.g., xy, x2 − y2) which describe their angular structure.
An orbital can be occupied by a maximum of two electrons, each with its own projection of spin . The simple names s orbital, p orbital, d orbital, and f orbital refer to orbitals with angular momentum quantum number ℓ = 0, 1, 2, and 3 respectively. These names, together with the value of n, are used to describe the electron configurations of atoms. They are derived from the description by early spectroscopists of certain series of alkali metal spectroscopic lines as sharp, principal, diffuse, and fundamental. Orbitals for ℓ > 3 continue alphabetically (g, h, i, k, ...),[3] omitting j[4][5] because some languages do not distinguish between the letters "i" and "j".[6]

Atomic orbitals are the basic building blocks of the atomic orbital model (or electron cloud or wave mechanics model), a modern framework for visualizing the submicroscopic behavior of electrons in matter. In this model the electron cloud of an atom may be seen as being built up (in approximation) in an electron configuration that is a product of simpler hydrogen-like atomic orbitals. The repeating periodicity of blocks of 2, 6, 10, and 14 elements within sections of the periodic table arises naturally from the total number of electrons that occupy a complete set of s, p, d, and f orbitals, respectively, though for higher values of quantum number n, particularly when the atom bears a positive charge, the energies of certain sub-shells become very similar and so the order in which they are said to be populated by electrons (e.g., Cr = [Ar]4s13d5 and Cr2+ = [Ar]3d4) can be rationalized only somewhat arbitrarily.

YouTube Encyclopedic
-
1/5Views:4 181 245793 5413 131 950231 698564 166
-
Quantum Numbers, Atomic Orbitals, and Electron Configurations
-
Orbitals, Atomic Energy Levels, & Sublevels Explained - Basic Introduction to Quantum Numbers
-
Orbitals: Crash Course Chemistry #25
-
What ARE atomic orbitals?
-
Atomic orbitals - electron configuration of Scandium (Z=21)
Transcription
Electron properties
With the development of quantum mechanics and experimental findings (such as the two slit diffraction of electrons), it was found that the electrons orbiting a nucleus could not be fully described as particles, but needed to be explained by wave–particle duality. In this sense, electrons have the following properties:
Wave-like properties:
- Electrons do not orbit a nucleus in the manner of a planet orbiting a star, but instead exist as standing waves. Thus the lowest possible energy an electron can take is similar to the fundamental frequency of a wave on a string. Higher energy states are similar to harmonics of that fundamental frequency.
- The electrons are never in a single point location, though the probability of interacting with the electron at a single point can be found from the electron's wave function. The electron's charge acts like it is smeared out in space in a continuous distribution, proportional at any point to the squared magnitude of the electron's wave function.
Particle-like properties:
- The number of electrons orbiting a nucleus can be only an integer.
- Electrons jump between orbitals like particles. For example, if one photon strikes the electrons, only one electron changes state as a result.
- Electrons retain particle-like properties such as: each wave state has the same electric charge as its electron particle. Each wave state has a single discrete spin (spin up or spin down) depending on its superposition.
Thus, electrons cannot be described simply as solid particles. An analogy might be that of a large and often oddly shaped "atmosphere" (the electron), distributed around a relatively tiny planet (the nucleus). Atomic orbitals exactly describe the shape of this "atmosphere" only when one electron is present. When more electrons are added, the additional electrons tend to more evenly fill in a volume of space around the nucleus so that the resulting collection ("electron cloud"[7]) tends toward a generally spherical zone of probability describing the electron's location, because of the uncertainty principle.
One should remember that these orbital 'states', as described here, are merely eigenstates of an electron in its orbit. An actual electron exists in a superposition of states, which is like a weighted average, but with complex number weights. So, for instance, an electron could be in a state (2, 1, 0), a pure eigenstate, or a mixed state ½(2, 1, 0) + ½(2, 1, 1), or even the mixed state ⅖(2, 1, 0) + ⅗ (2, 1, 1). For each eigenstate, a property has an eigenvalue. So, for the three states just mentioned, the value of is 2, and the value of is 1. But the value for is a superposition of 0 and 1. It is not a fraction like ½; it is ambiguous, either 0.0 or 1.0. A superposition of eigenstates (2, 1, 1) and (3, 2, 1) would have an ambiguous and , but would definitely be 1. Eigenstates make it easier to deal with the math. You can choose a different basis of eigenstates by superimposing eigenstates from any other basis (see Real orbitals below).




Formal quantum mechanical definition
Atomic orbitals may be defined more precisely in formal quantum mechanical language. They are approximate solutions to the Schrödinger equation for the electrons bound to the atom by the electric field of the atom's nucleus. Specifically, in quantum mechanics, the state of an atom, i.e., an eigenstate of the atomic Hamiltonian, is approximated by an expansion (see configuration interaction expansion and basis set) into linear combinations of anti-symmetrized products (Slater determinants) of one-electron functions. The spatial components of these one-electron functions are called atomic orbitals. (When one considers also their spin component, one speaks of atomic spin orbitals.) A state is actually a function of the coordinates of all the electrons, so that their motion is correlated, but this is often approximated by this independent-particle model of products of single electron wave functions.[8] (The London dispersion force, for example, depends on the correlations of the motion of the electrons.)
In atomic physics, the atomic spectral lines correspond to transitions (quantum leaps) between quantum states of an atom. These states are labeled by a set of quantum numbers summarized in the term symbol and usually associated with particular electron configurations, i.e., by occupation schemes of atomic orbitals (for example, 1s2 2s2 2p6 for the ground state of neon-term symbol: 1S0).
This notation means that the corresponding Slater determinants have a clear higher weight in the configuration interaction expansion. The atomic orbital concept is therefore a key concept for visualizing the excitation process associated with a given transition. For example, one can say for a given transition that it corresponds to the excitation of an electron from an occupied orbital to a given unoccupied orbital. Nevertheless, one has to keep in mind that electrons are fermions ruled by the Pauli exclusion principle and cannot be distinguished from each other.[9] Moreover, it sometimes happens that the configuration interaction expansion converges very slowly and that one cannot speak about simple one-determinant wave function at all. This is the case when electron correlation is large.
Fundamentally, an atomic orbital is a one-electron wave function, even though many electrons are not in one-electron atoms, and so the one-electron view is an approximation. When thinking about orbitals, we are often given an orbital visualization heavily influenced by the Hartree–Fock approximation, which is one way to reduce the complexities of molecular orbital theory.
Types of orbital
Atomic orbitals can be the hydrogen-like "orbitals" which are exact solutions to the Schrödinger equation for a hydrogen-like "atom" (i.e., atom with one electron). Alternatively, atomic orbitals refer to functions that depend on the coordinates of one electron (i.e., orbitals) but are used as starting points for approximating wave functions that depend on the simultaneous coordinates of all the electrons in an atom or molecule. The coordinate systems chosen for orbitals are usually spherical coordinates (r, θ, φ) in atoms and Cartesian (x, y, z) in polyatomic molecules. The advantage of spherical coordinates here is that an orbital wave function is a product of three factors each dependent on a single coordinate: ψ(r, θ, φ) = R(r) Θ(θ) Φ(φ). The angular factors of atomic orbitals Θ(θ) Φ(φ) generate s, p, d, etc. functions as real combinations of spherical harmonics Yℓm(θ, φ) (where ℓ and m are quantum numbers). There are typically three mathematical forms for the radial functions R(r) which can be chosen as a starting point for the calculation of the properties of atoms and molecules with many electrons:
- The hydrogen-like orbitals are derived from the exact solutions of the Schrödinger equation for one electron and a nucleus, for a hydrogen-like atom. The part of the function that depends on distance r from the nucleus has radial nodes and decays as .
- The Slater-type orbital (STO) is a form without radial nodes but decays from the nucleus as does a hydrogen-like orbital.
- The form of the Gaussian type orbital (Gaussians) has no radial nodes and decays as .


Although hydrogen-like orbitals are still used as pedagogical tools, the advent of computers has made STOs preferable for atoms and diatomic molecules since combinations of STOs can replace the nodes in hydrogen-like orbitals. Gaussians are typically used in molecules with three or more atoms. Although not as accurate by themselves as STOs, combinations of many Gaussians can attain the accuracy of hydrogen-like orbitals.
History
The term "orbital" was coined by Robert S. Mulliken in 1932 as short for one-electron orbital wave function.[10] Niels Bohr explained around 1913 that electrons might revolve around a compact nucleus with definite angular momentum.[11] Bohr's model was an improvement on the 1911 explanations of Ernest Rutherford, that of the electron moving around a nucleus. Japanese physicist Hantaro Nagaoka published an orbit-based hypothesis for electron behavior as early as 1904.[12] These theories were each built upon new observations starting with simple understanding and becoming more correct and complex. Explaining the behavior of these electron "orbits" was one of the driving forces behind the development of quantum mechanics.[13]
Early models
With J. J. Thomson's discovery of the electron in 1897,[14] it became clear that atoms were not the smallest building blocks of nature, but were rather composite particles. The newly discovered structure within atoms tempted many to imagine how the atom's constituent parts might interact with each other. Thomson theorized that multiple electrons revolve in orbit-like rings within a positively charged jelly-like substance,[15] and between the electron's discovery and 1909, this "plum pudding model" was the most widely accepted explanation of atomic structure.
Shortly after Thomson's discovery, Hantaro Nagaoka predicted a different model for electronic structure.[12] Unlike the plum pudding model, the positive charge in Nagaoka's "Saturnian Model" was concentrated into a central core, pulling the electrons into circular orbits reminiscent of Saturn's rings. Few people took notice of Nagaoka's work at the time,[16] and Nagaoka himself recognized a fundamental defect in the theory even at its conception, namely that a classical charged object cannot sustain orbital motion because it is accelerating and therefore loses energy due to electromagnetic radiation.[17] Nevertheless, the Saturnian model turned out to have more in common with modern theory than any of its contemporaries.
Bohr atom
In 1909, Ernest Rutherford discovered that the bulk of the atomic mass was tightly condensed into a nucleus, which was also found to be positively charged. It became clear from his analysis in 1911 that the plum pudding model could not explain atomic structure. In 1913, Rutherford's post-doctoral student, Niels Bohr, proposed a new model of the atom, wherein electrons orbited the nucleus with classical periods, but were permitted to have only discrete values of angular momentum, quantized in units ħ.[11] This constraint automatically allowed only certain electron energies. The Bohr model of the atom fixed the problem of energy loss from radiation from a ground state (by declaring that there was no state below this), and more importantly explained the origin of spectral lines.

After Bohr's use of Einstein's explanation of the photoelectric effect to relate energy levels in atoms with the wavelength of emitted light, the connection between the structure of electrons in atoms and the emission and absorption spectra of atoms became an increasingly useful tool in the understanding of electrons in atoms. The most prominent feature of emission and absorption spectra (known experimentally since the middle of the 19th century), was that these atomic spectra contained discrete lines. The significance of the Bohr model was that it related the lines in emission and absorption spectra to the energy differences between the orbits that electrons could take around an atom. This was, however, not achieved by Bohr through giving the electrons some kind of wave-like properties, since the idea that electrons could behave as matter waves was not suggested until eleven years later. Still, the Bohr model's use of quantized angular momenta and therefore quantized energy levels was a significant step toward the understanding of electrons in atoms, and also a significant step towards the development of quantum mechanics in suggesting that quantized restraints must account for all discontinuous energy levels and spectra in atoms.
With de Broglie's suggestion of the existence of electron matter waves in 1924, and for a short time before the full 1926 Schrödinger equation treatment of hydrogen-like atoms, a Bohr electron "wavelength" could be seen to be a function of its momentum; so a Bohr orbiting electron was seen to orbit in a circle at a multiple of its half-wavelength. The Bohr model for a short time could be seen as a classical model with an additional constraint provided by the 'wavelength' argument. However, this period was immediately superseded by the full three-dimensional wave mechanics of 1926. In our current understanding of physics, the Bohr model is called a semi-classical model because of its quantization of angular momentum, not primarily because of its relationship with electron wavelength, which appeared in hindsight a dozen years after the Bohr model was proposed.
The Bohr model was able to explain the emission and absorption spectra of hydrogen. The energies of electrons in the n = 1, 2, 3, etc. states in the Bohr model match those of current physics. However, this did not explain similarities between different atoms, as expressed by the periodic table, such as the fact that helium (two electrons), neon (10 electrons), and argon (18 electrons) exhibit similar chemical inertness. Modern quantum mechanics explains this in terms of electron shells and subshells which can each hold a number of electrons determined by the Pauli exclusion principle. Thus the n = 1 state can hold one or two electrons, while the n = 2 state can hold up to eight electrons in 2s and 2p subshells. In helium, all n = 1 states are fully occupied; the same is true for n = 1 and n = 2 in neon. In argon, the 3s and 3p subshells are similarly fully occupied by eight electrons; quantum mechanics also allows a 3d subshell but this is at higher energy than the 3s and 3p in argon (contrary to the situation for hydrogen) and remains empty.
Modern conceptions and connections to the Heisenberg uncertainty principle
Immediately after Heisenberg discovered his uncertainty principle,[18] Bohr noted that the existence of any sort of wave packet implies uncertainty in the wave frequency and wavelength, since a spread of frequencies is needed to create the packet itself.[19] In quantum mechanics, where all particle momenta are associated with waves, it is the formation of such a wave packet which localizes the wave, and thus the particle, in space. In states where a quantum mechanical particle is bound, it must be localized as a wave packet, and the existence of the packet and its minimum size implies a spread and minimal value in particle wavelength, and thus also momentum and energy. In quantum mechanics, as a particle is localized to a smaller region in space, the associated compressed wave packet requires a larger and larger range of momenta, and thus larger kinetic energy. Thus the binding energy to contain or trap a particle in a smaller region of space increases without bound as the region of space grows smaller. Particles cannot be restricted to a geometric point in space, since this would require infinite particle momentum.
In chemistry, Erwin Schrödinger, Linus Pauling, Mulliken and others noted that the consequence of Heisenberg's relation was that the electron, as a wave packet, could not be considered to have an exact location in its orbital. Max Born suggested that the electron's position needed to be described by a probability distribution which was connected with finding the electron at some point in the wave-function which described its associated wave packet. The new quantum mechanics did not give exact results, but only the probabilities for the occurrence of a variety of possible such results. Heisenberg held that the path of a moving particle has no meaning if we cannot observe it, as we cannot with electrons in an atom.
In the quantum picture of Heisenberg, Schrödinger and others, the Bohr atom number n for each orbital became known as an n-sphere[citation needed] in a three-dimensional atom and was pictured as the most probable energy of the probability cloud of the electron's wave packet which surrounded the atom.
Orbital names
Orbital notation and subshells
Orbitals have been given names, which are usually given in the form:

where X is the energy level corresponding to the principal quantum number n; type is a lower-case letter denoting the shape or subshell of the orbital, corresponding to the angular momentum quantum number ℓ.
For example, the orbital 1s (pronounced as the individual numbers and letters: "'one' 'ess'") is the lowest energy level (n = 1) and has an angular quantum number of ℓ = 0, denoted as s. Orbitals with ℓ = 1, 2 and 3 are denoted as p, d and f respectively.
The set of orbitals for a given n and ℓ is called a subshell, denoted
- .

The superscript y shows the number of electrons in the subshell. For example, the notation 2p4 indicates that the 2p subshell of an atom contains 4 electrons. This subshell has 3 orbitals, each with n = 2 and ℓ = 1.
X-ray notation
There is also another, less common system still used in X-ray science known as X-ray notation, which is a continuation of the notations used before orbital theory was well understood. In this system, the principal quantum number is given a letter associated with it. For n = 1, 2, 3, 4, 5, ..., the letters associated with those numbers are K, L, M, N, O, ... respectively.
Hydrogen-like orbitals
The simplest atomic orbitals are those that are calculated for systems with a single electron, such as the hydrogen atom. An atom of any other element ionized down to a single electron is very similar to hydrogen, and the orbitals take the same form. In the Schrödinger equation for this system of one negative and one positive particle, the atomic orbitals are the eigenstates of the Hamiltonian operator for the energy. They can be obtained analytically, meaning that the resulting orbitals are products of a polynomial series, and exponential and trigonometric functions. (see hydrogen atom).
For atoms with two or more electrons, the governing equations can be solved only with the use of methods of iterative approximation. Orbitals of multi-electron atoms are qualitatively similar to those of hydrogen, and in the simplest models, they are taken to have the same form. For more rigorous and precise analysis, numerical approximations must be used.
A given (hydrogen-like) atomic orbital is identified by unique values of three quantum numbers: n, ℓ, and mℓ. The rules restricting the values of the quantum numbers, and their energies (see below), explain the electron configuration of the atoms and the periodic table.
The stationary states (quantum states) of the hydrogen-like atoms are its atomic orbitals.[clarification needed] However, in general, an electron's behavior is not fully described by a single orbital. Electron states are best represented by time-depending "mixtures" (linear combinations) of multiple orbitals. See Linear combination of atomic orbitals molecular orbital method.
The quantum number n first appeared in the Bohr model where it determines the radius of each circular electron orbit. In modern quantum mechanics however, n determines the mean distance of the electron from the nucleus; all electrons with the same value of n lie at the same average distance. For this reason, orbitals with the same value of n are said to comprise a "shell". Orbitals with the same value of n and also the same value of ℓ are even more closely related, and are said to comprise a "subshell".
Quantum numbers
Because of the quantum mechanical nature of the electrons around a nucleus, atomic orbitals can be uniquely defined by a set of integers known as quantum numbers. These quantum numbers occur only in certain combinations of values, and their physical interpretation changes depending on whether real or complex versions of the atomic orbitals are employed.
Complex orbitals
In physics, the most common orbital descriptions are based on the solutions to the hydrogen atom, where orbitals are given by the product between a radial function and a pure spherical harmonic. The quantum numbers, together with the rules governing their possible values, are as follows:
The principal quantum number n describes the energy of the electron and is always a positive integer. In fact, it can be any positive integer, but for reasons discussed below, large numbers are seldom encountered. Each atom has, in general, many orbitals associated with each value of n; these orbitals together are sometimes called electron shells.
The azimuthal quantum number ℓ describes the orbital angular momentum of each electron and is a non-negative integer. Within a shell where n is some integer n0, ℓ ranges across all (integer) values satisfying the relation . For instance, the n = 1 shell has only orbitals with , and the n = 2 shell has only orbitals with , and . The set of orbitals associated with a particular value of ℓ are sometimes collectively called a subshell.



The magnetic quantum number, , describes the projection of the orbital angular momentum along a chosen axis. It determines the magnitude of the current circulating around that axis and the orbital contribution to the magnetic moment of an electron via the Ampèrian loop model.[20] Within a subshell , obtains the integer values in the range .



The above results may be summarized in the following table. Each cell represents a subshell, and lists the values of available in that subshell. Empty cells represent subshells that do not exist.
| ℓ = 0 (s) | ℓ = 1 (p) | ℓ = 2 (d) | ℓ = 3 (f) | ℓ = 4 (g) | ... | |
|---|---|---|---|---|---|---|
| n = 1 | ... | |||||
| n = 2 | 0 | −1, 0, 1 | ... | |||
| n = 3 | 0 | −1, 0, 1 | −2, −1, 0, 1, 2 | ... | ||
| n = 4 | 0 | −1, 0, 1 | −2, −1, 0, 1, 2 | −3, −2, −1, 0, 1, 2, 3 | ... | |
| n = 5 | 0 | −1, 0, 1 | −2, −1, 0, 1, 2 | −3, −2, −1, 0, 1, 2, 3 | −4, −3, −2, −1, 0, 1, 2, 3, 4 | ... |
| ... | ... | ... | ... | ... | ... | ... |

Subshells are usually identified by their - and -values. is represented by its numerical value, but is represented by a letter as follows: 0 is represented by 's', 1 by 'p', 2 by 'd', 3 by 'f', and 4 by 'g'. For instance, one may speak of the subshell with and as a '2s subshell'.

Each electron also has angular momentum in the form of quantum mechanical spin given by spin s = 1/2. Its projection along a specified axis is given by the spin magnetic quantum number, ms, which can be +1/2 or −1/2. These values are also called "spin up" or "spin down" respectively.
The Pauli exclusion principle states that no two electrons in an atom can have the same values of all four quantum numbers. If there are two electrons in an orbital with given values for three quantum numbers, (n, ℓ, m), these two electrons must differ in their spin projection ms.
The above conventions imply a preferred axis (for example, the z direction in Cartesian coordinates), and they also imply a preferred direction along this preferred axis. Otherwise there would be no sense in distinguishing m = +1 from m = −1. As such, the model is most useful when applied to physical systems that share these symmetries. The Stern–Gerlach experiment—where an atom is exposed to a magnetic field—provides one such example.[21]
Real orbitals
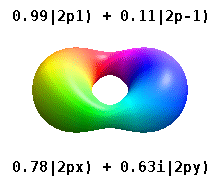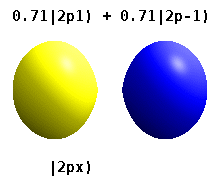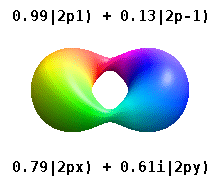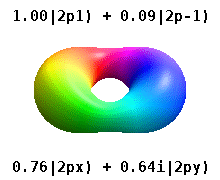


Instead of the complex orbitals described above, it is common, especially in the chemistry literature, to use real atomic orbitals. These real orbitals arise from simple linear combinations of complex orbitals. Using the Condon–Shortley phase convention, real orbitals are related to complex orbitals in the same way that the real spherical harmonics are related to complex spherical harmonics. Letting denote a complex orbital with quantum numbers , , and , the real orbitals may be defined by[22]




If , with the radial part of the orbital, this definition is equivalent to where is the real spherical harmonic related to either the real or imaginary part of the complex spherical harmonic .





Real spherical harmonics are physically relevant when an atom is embedded in a crystalline solid, in which case there are multiple preferred symmetry axes but no single preferred direction.[citation needed] Real atomic orbitals are also more frequently encountered in introductory chemistry textbooks and shown in common orbital visualizations.[23] In real hydrogen-like orbitals, quantum numbers and have the same interpretation and significance as their complex counterparts, but is no longer a good quantum number (but its absolute value is).
Some real orbitals are given specific names beyond the simple designation. Orbitals with quantum number equal to are called orbitals. With this one can already assign names to complex orbitals such as ; the first symbol is the quantum number, the second character is the symbol for that particular quantum number and the subscript is the quantum number.



As an example of how the full orbital names are generated for real orbitals, one may calculate . From the table of spherical harmonics, with . Then




Likewise . As a more complicated example:


In all these cases we generate a Cartesian label for the orbital by examining, and abbreviating, the polynomial in , , and appearing in the numerator. We ignore any terms in the polynomial except for the term with the highest exponent in . We then use the abbreviated polynomial as a subscript label for the atomic state, using the same nomenclature as above to indicate the and quantum numbers.[citation needed]





The expression above all use the Condon–Shortley phase convention which is favored by quantum physicists.[24][25] Other conventions exist for the phase of the spherical harmonics.[26][27] Under these different conventions the and orbitals may appear, for example, as the sum and difference of and , contrary to what is shown above.



Below is a list of these Cartesian polynomial names for the atomic orbitals.[28][29] There does not seem to be reference in the literature as to how to abbreviate the long Cartesian spherical harmonic polynomials for so there does not seem be consensus on the naming of orbitals or higher according to this nomenclature.[citation needed]

























Shapes of orbitals

Simple pictures showing orbital shapes are intended to describe the angular forms of regions in space where the electrons occupying the orbital are likely to be found. The diagrams cannot show the entire region where an electron can be found, since according to quantum mechanics there is a non-zero probability of finding the electron (almost) anywhere in space. Instead the diagrams are approximate representations of boundary or contour surfaces where the probability density | ψ(r, θ, φ) |2 has a constant value, chosen so that there is a certain probability (for example 90%) of finding the electron within the contour. Although | ψ |2 as the square of an absolute value is everywhere non-negative, the sign of the wave function ψ(r, θ, φ) is often indicated in each subregion of the orbital picture.
Sometimes the ψ function is graphed to show its phases, rather than | ψ(r, θ, φ) |2 which shows probability density but has no phase (which is lost when taking absolute value, since ψ(r, θ, φ) is a complex number). |ψ(r, θ, φ)|2 orbital graphs tend to have less spherical, thinner lobes than ψ(r, θ, φ) graphs, but have the same number of lobes in the same places, and otherwise are recognizable. This article, to show wave function phase, shows mostly ψ(r, θ, φ) graphs.
The lobes can be seen as standing wave interference patterns between the two counter-rotating, ring-resonant traveling wave m and −m modes; the projection of the orbital onto the xy plane has a resonant m wavelength around the circumference. Although rarely shown, the traveling wave solutions can be seen as rotating banded tori; the bands represent phase information. For each m there are two standing wave solutions ⟨m⟩ + ⟨−m⟩ and ⟨m⟩ − ⟨−m⟩. If m = 0, the orbital is vertical, counter rotating information is unknown, and the orbital is z-axis symmetric. If ℓ = 0 there are no counter rotating modes. There are only radial modes and the shape is spherically symmetric.
Nodal planes and nodal spheres are surfaces on which the probability density vanishes. The number of nodal surfaces is controlled by the quantum numbers n and ℓ. An orbital with azimuthal quantum number ℓ has ℓ radial nodal planes passing through the origin. For example, the s orbitals (ℓ = 0) are spherically symmetric and have no nodal planes, whereas the p orbitals (ℓ = 1) have a single nodal plane between the lobes. The number of nodal spheres equals n-ℓ-1, consistent with the restriction ℓ ≤ n-1 on the quantum numbers. The principal quantum number controls the total number of nodal surfaces which is n-1.[30] Loosely speaking, n is energy, ℓ is analogous to eccentricity, and m is orientation.
In general, n determines size and energy of the orbital for a given nucleus; as n increases, the size of the orbital increases. The higher nuclear charge Z of heavier elements causes their orbitals to contract by comparison to lighter ones, so that the size of the atom remains very roughly constant, even as the number of electrons increases.

Also in general terms, ℓ determines an orbital's shape, and mℓ its orientation. However, since some orbitals are described by equations in complex numbers, the shape sometimes depends on mℓ also. Together, the whole set of orbitals for a given ℓ and n fill space as symmetrically as possible, though with increasingly complex sets of lobes and nodes.
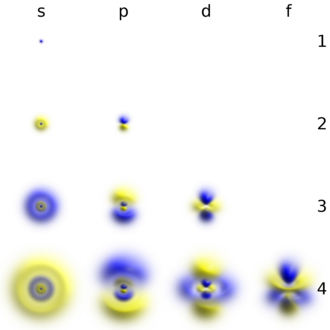
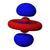
The single s orbitals () are shaped like spheres. For n = 1 it is roughly a solid ball (densest at center and fades outward exponentially), but for n ≥ 2, each single s orbital is made of spherically symmetric surfaces which are nested shells (i.e., the "wave-structure" is radial, following a sinusoidal radial component as well). See illustration of a cross-section of these nested shells, at right. The s orbitals for all n numbers are the only orbitals with an anti-node (a region of high wave function density) at the center of the nucleus. All other orbitals (p, d, f, etc.) have angular momentum, and thus avoid the nucleus (having a wave node at the nucleus). Recently, there has been an effort to experimentally image the 1s and 2p orbitals in a SrTiO3 crystal using scanning transmission electron microscopy with energy dispersive x-ray spectroscopy.[31] Because the imaging was conducted using an electron beam, Coulombic beam-orbital interaction that is often termed as the impact parameter effect is included in the outcome (see the figure at right).
The shapes of p, d and f orbitals are described verbally here and shown graphically in the Orbitals table below. The three p orbitals for n = 2 have the form of two ellipsoids with a point of tangency at the nucleus (the two-lobed shape is sometimes referred to as a "dumbbell"—there are two lobes pointing in opposite directions from each other). The three p orbitals in each shell are oriented at right angles to each other, as determined by their respective linear combination of values of mℓ. The overall result is a lobe pointing along each direction of the primary axes.
Four of the five d orbitals for n = 3 look similar, each with four pear-shaped lobes, each lobe tangent at right angles to two others, and the centers of all four lying in one plane. Three of these planes are the xy-, xz-, and yz-planes—the lobes are between the pairs of primary axes—and the fourth has the center along the x and y axes themselves. The fifth and final d orbital consists of three regions of high probability density: a torus in between two pear-shaped regions placed symmetrically on its z axis. The overall total of 18 directional lobes point in every primary axis direction and between every pair.
There are seven f orbitals, each with shapes more complex than those of the d orbitals.
Additionally, as is the case with the s orbitals, individual p, d, f and g orbitals with n values higher than the lowest possible value, exhibit an additional radial node structure which is reminiscent of harmonic waves of the same type, as compared with the lowest (or fundamental) mode of the wave. As with s orbitals, this phenomenon provides p, d, f, and g orbitals at the next higher possible value of n (for example, 3p orbitals vs. the fundamental 2p), an additional node in each lobe. Still higher values of n further increase the number of radial nodes, for each type of orbital.
The shapes of atomic orbitals in one-electron atom are related to 3-dimensional spherical harmonics. These shapes are not unique, and any linear combination is valid, like a transformation to cubic harmonics, in fact it is possible to generate sets where all the d's are the same shape, just like the px, py, and pz are the same shape.[32][33]

Although individual orbitals are most often shown independent of each other, the orbitals coexist around the nucleus at the same time. Also, in 1927, Albrecht Unsöld proved that if one sums the electron density of all orbitals of a particular azimuthal quantum number ℓ of the same shell n (e.g., all three 2p orbitals, or all five 3d orbitals) where each orbital is occupied by an electron or each is occupied by an electron pair, then all angular dependence disappears; that is, the resulting total density of all the atomic orbitals in that subshell (those with the same ℓ) is spherical. This is known as Unsöld's theorem.
Orbitals table
This table shows the real hydrogen-like wave functions for all atomic orbitals up to 7s, and therefore covers the occupied orbitals in the ground state of all elements in the periodic table up to radium and some beyond. "ψ" graphs are shown with − and + wave function phases shown in two different colors (arbitrarily red and blue). The pz orbital is the same as the p0 orbital, but the px and py are formed by taking linear combinations of the p+1 and p−1 orbitals (which is why they are listed under the m = ±1 label). Also, the p+1 and p−1 are not the same shape as the p0, since they are pure spherical harmonics.
| s (ℓ = 0) | p (ℓ = 1) | d (ℓ = 2) | f (ℓ = 3) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m = 0 | m = 0 | m = ±1 | m = 0 | m = ±1 | m = ±2 | m = 0 | m = ±1 | m = ±2 | m = ±3 | |||||||
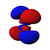
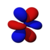
| s | pz | px | py | dz2 | dxz | dyz | dxy | dx2−y2 | fz3 | fxz2 | fyz2 | fxyz | fz(x2−y2) | fx(x2−3y2) | fy(3x2−y2) | |
| n = 1 | 
|
|||||||||||||||
| n = 2 | 
|

|

|

|
||||||||||||
| n = 3 | 
|

|

|

|

|

|

|

|
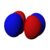
|
|||||||
| n = 4 | 
|

|

|

|
|
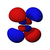
|
|

|

|

|
|

|

|

|

|

|
| n = 5 | 
|

|
|

|

|

|

|

|

|
. . . | . . . | . . . | . . . | . . . | . . . | . . . |
| n = 6 | 
|

|

|

|
. . . ‡ | . . . | . . . ‡ | . . . | . . . ‡ | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * |
| n = 7 | 
|
. . . † | . . . † | . . . † | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * | . . . * |
* No elements with 6f, 7d or 7f electrons have been discovered yet.
† Elements with 7p electrons have been discovered, but their electronic configurations are only predicted.
‡ For the elements whose highest occupied orbital is a 6d orbital, only some electronic configurations have been confirmed. (Mt, Ds, Rg and Cn are still missing).
These are the real-valued orbitals commonly used in chemistry. Only the orbitals where are eigenstates of the orbital angular momentum operator, . The columns with are combinations of two eigenstates. See comparison in the following picture:




Qualitative understanding of shapes
The shapes of atomic orbitals can be qualitatively understood by considering the analogous case of standing waves on a circular drum.[34] To see the analogy, the mean vibrational displacement of each bit of drum membrane from the equilibrium point over many cycles (a measure of average drum membrane velocity and momentum at that point) must be considered relative to that point's distance from the center of the drum head. If this displacement is taken as being analogous to the probability of finding an electron at a given distance from the nucleus, then it will be seen that the many modes of the vibrating disk form patterns that trace the various shapes of atomic orbitals. The basic reason for this correspondence lies in the fact that the distribution of kinetic energy and momentum in a matter-wave is predictive of where the particle associated with the wave will be. That is, the probability of finding an electron at a given place is also a function of the electron's average momentum at that point, since high electron momentum at a given position tends to "localize" the electron in that position, via the properties of electron wave-packets (see the Heisenberg uncertainty principle for details of the mechanism).
This relationship means that certain key features can be observed in both drum membrane modes and atomic orbitals. For example, in all of the modes analogous to s orbitals (the top row in the animated illustration below), it can be seen that the very center of the drum membrane vibrates most strongly, corresponding to the antinode in all s orbitals in an atom. This antinode means the electron is most likely to be at the physical position of the nucleus (which it passes straight through without scattering or striking it), since it is moving (on average) most rapidly at that point, giving it maximal momentum.
A mental "planetary orbit" picture closest to the behavior of electrons in s orbitals, all of which have no angular momentum, might perhaps be that of a Keplerian orbit with the orbital eccentricity of 1 but a finite major axis, not physically possible (because particles were to collide), but can be imagined as a limit of orbits with equal major axes but increasing eccentricity.
Below, a number of drum membrane vibration modes and the respective wave functions of the hydrogen atom are shown. A correspondence can be considered where the wave functions of a vibrating drum head are for a two-coordinate system ψ(r, θ) and the wave functions for a vibrating sphere are three-coordinate ψ(r, θ, φ).
- s-type drum modes and wave functions
-
 Drum mode
Drum mode -
 Drum mode
Drum mode -
 Drum mode
Drum mode -
 Wave function of 1s orbital (real part, 2D-cut, )
Wave function of 1s orbital (real part, 2D-cut, ) -
 Wave function of 2s orbital (real part, 2D-cut, )
Wave function of 2s orbital (real part, 2D-cut, ) -
 Wave function of 3s orbital (real part, 2D-cut, )
Wave function of 3s orbital (real part, 2D-cut, )












None of the other sets of modes in a drum membrane have a central antinode, and in all of them the center of the drum does not move. These correspond to a node at the nucleus for all non-s orbitals in an atom. These orbitals all have some angular momentum, and in the planetary model, they correspond to particles in orbit with eccentricity less than 1.0, so that they do not pass straight through the center of the primary body, but keep somewhat away from it.
In addition, the drum modes analogous to p and d modes in an atom show spatial irregularity along the different radial directions from the center of the drum, whereas all of the modes analogous to s modes are perfectly symmetrical in radial direction. The non-radial-symmetry properties of non-s orbitals are necessary to localize a particle with angular momentum and a wave nature in an orbital where it must tend to stay away from the central attraction force, since any particle localized at the point of central attraction could have no angular momentum. For these modes, waves in the drum head tend to avoid the central point. Such features again emphasize that the shapes of atomic orbitals are a direct consequence of the wave nature of electrons.
- p-type drum modes and wave functions
-
 Drum mode
Drum mode -
 Drum mode
Drum mode -
 Drum mode
Drum mode -
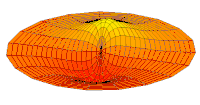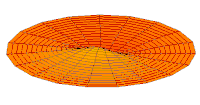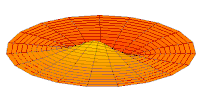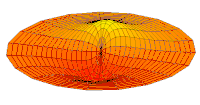 Wave function of 2p orbital (real part, 2D-cut, )
Wave function of 2p orbital (real part, 2D-cut, ) -
 Wave function of 3p orbital (real part, 2D-cut, )
Wave function of 3p orbital (real part, 2D-cut, ) -
 Wave function of 4p orbital (real part, 2D-cut, )
Wave function of 4p orbital (real part, 2D-cut, )










- d-type drum modes
-
 Drum mode
Drum mode -
 Drum mode
Drum mode -
 Drum mode
Drum mode






Orbital energy
In atoms with one electron (hydrogen-like atom), the energy of an orbital (and, consequently, any electron in the orbital) is determined mainly by . The orbital has the lowest possible energy in the atom. Each successively higher value of has a higher energy, but the difference decreases as increases. For high , the energy becomes so high that the electron can easily escape the atom. In single electron atoms, all levels with different within a given are degenerate in the Schrödinger approximation, and have the same energy. This approximation is broken slightly in the solution to the Dirac equation (where energy depends on n and another quantum number j), and by the effect of the magnetic field of the nucleus and quantum electrodynamics effects. The latter induce tiny binding energy differences especially for s electrons that go nearer the nucleus, since these feel a very slightly different nuclear charge, even in one-electron atoms; see Lamb shift.

In atoms with multiple electrons, the energy of an electron depends not only on its orbital, but also on its interactions with other electrons. These interactions depend on the detail of its spatial probability distribution, and so the energy levels of orbitals depend not only on but also on . Higher values of are associated with higher values of energy; for instance, the 2p state is higher than the 2s state. When , the increase in energy of the orbital becomes so large as to push the energy of orbital above the energy of the s orbital in the next higher shell; when the energy is pushed into the shell two steps higher. The filling of the 3d orbitals does not occur until the 4s orbitals have been filled.
The increase in energy for subshells of increasing angular momentum in larger atoms is due to electron–electron interaction effects, and it is specifically related to the ability of low angular momentum electrons to penetrate more effectively toward the nucleus, where they are subject to less screening from the charge of intervening electrons. Thus, in atoms with higher atomic number, the of electrons becomes more and more of a determining factor in their energy, and the principal quantum numbers of electrons becomes less and less important in their energy placement.
The energy sequence of the first 35 subshells (e.g., 1s, 2p, 3d, etc.) is given in the following table. Each cell represents a subshell with and given by its row and column indices, respectively. The number in the cell is the subshell's position in the sequence. For a linear listing of the subshells in terms of increasing energies in multielectron atoms, see the section below.
l n
|
s | p | d | f | g | h |
|---|---|---|---|---|---|---|
| 1 | 1 | |||||
| 2 | 2 | 3 | ||||
| 3 | 4 | 5 | 7 | |||
| 4 | 6 | 8 | 10 | 13 | ||
| 5 | 9 | 11 | 14 | 17 | 21 | |
| 6 | 12 | 15 | 18 | 22 | 26 | 31 |
| 7 | 16 | 19 | 23 | 27 | 32 | 37 |
| 8 | 20 | 24 | 28 | 33 | 38 | 44 |
| 9 | 25 | 29 | 34 | 39 | 45 | 51 |
| 10 | 30 | 35 | 40 | 46 | 52 | 59 |
Note: empty cells indicate non-existent sublevels, while numbers in italics indicate sublevels that could (potentially) exist, but which do not hold electrons in any element currently known.
Electron placement and the periodic table

Several rules govern the placement of electrons in orbitals (electron configuration). The first dictates that no two electrons in an atom may have the same set of values of quantum numbers (this is the Pauli exclusion principle). These quantum numbers include the three that define orbitals, as well as the spin magnetic quantum number ms. Thus, two electrons may occupy a single orbital, so long as they have different values of ms. Because ms takes one of only two values (1/2 or -1/2), at most two electrons can occupy each orbital.
Additionally, an electron always tends to fall to the lowest possible energy state. It is possible for it to occupy any orbital so long as it does not violate the Pauli exclusion principle, but if lower-energy orbitals are available, this condition is unstable. The electron will eventually lose energy (by releasing a photon) and drop into the lower orbital. Thus, electrons fill orbitals in the order specified by the energy sequence given above.
This behavior is responsible for the structure of the periodic table. The table may be divided into several rows (called 'periods'), numbered starting with 1 at the top. The presently known elements occupy seven periods. If a certain period has number i, it consists of elements whose outermost electrons fall in the ith shell. Niels Bohr was the first to propose (1923) that the periodicity in the properties of the elements might be explained by the periodic filling of the electron energy levels, resulting in the electronic structure of the atom.[35]
The periodic table may also be divided into several numbered rectangular 'blocks'. The elements belonging to a given block have this common feature: their highest-energy electrons all belong to the same ℓ-state (but the n associated with that ℓ-state depends upon the period). For instance, the leftmost two columns constitute the 's-block'. The outermost electrons of Li and Be respectively belong to the 2s subshell, and those of Na and Mg to the 3s subshell.
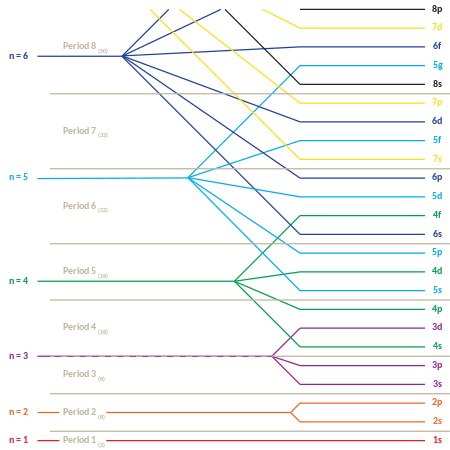
The following is the order for filling the "subshell" orbitals, which also gives the order of the "blocks" in the periodic table:
- 1s, 2s, 2p, 3s, 3p, 4s, 3d, 4p, 5s, 4d, 5p, 6s, 4f, 5d, 6p, 7s, 5f, 6d, 7p
The "periodic" nature of the filling of orbitals, as well as emergence of the s, p, d, and f "blocks", is more obvious if this order of filling is given in matrix form, with increasing principal quantum numbers starting the new rows ("periods") in the matrix. Then, each subshell (composed of the first two quantum numbers) is repeated as many times as required for each pair of electrons it may contain. The result is a compressed periodic table, with each entry representing two successive elements:
| 1s | |||||||||||||||
| 2s | 2p | 2p | 2p | ||||||||||||
| 3s | 3p | 3p | 3p | ||||||||||||
| 4s | 3d | 3d | 3d | 3d | 3d | 4p | 4p | 4p | |||||||
| 5s | 4d | 4d | 4d | 4d | 4d | 5p | 5p | 5p | |||||||
| 6s | 4f | 4f | 4f | 4f | 4f | 4f | 4f | 5d | 5d | 5d | 5d | 5d | 6p | 6p | 6p |
| 7s | 5f | 5f | 5f | 5f | 5f | 5f | 5f | 6d | 6d | 6d | 6d | 6d | 7p | 7p | 7p |
Although this is the general order of orbital filling according to the Madelung rule, there are exceptions, and the actual electronic energies of each element are also dependent upon additional details of the atoms (see Electron configuration § Atoms: Aufbau principle and Madelung rule).
The number of electrons in an electrically neutral atom increases with the atomic number. The electrons in the outermost shell, or valence electrons, tend to be responsible for an element's chemical behavior. Elements that contain the same number of valence electrons can be grouped together and display similar chemical properties.
Relativistic effects
For elements with high atomic number Z, the effects of relativity become more pronounced, and especially so for s electrons, which move at relativistic velocities as they penetrate the screening electrons near the core of high-Z atoms. This relativistic increase in momentum for high speed electrons causes a corresponding decrease in wavelength and contraction of 6s orbitals relative to 5d orbitals (by comparison to corresponding s and d electrons in lighter elements in the same column of the periodic table); this results in 6s valence electrons becoming lowered in energy.
Examples of significant physical outcomes of this effect include the lowered melting temperature of mercury (which results from 6s electrons not being available for metal bonding) and the golden color of gold and caesium.[36]
In the Bohr model, an n = 1 electron has a velocity given by , where Z is the atomic number, is the fine-structure constant, and c is the speed of light. In non-relativistic quantum mechanics, therefore, any atom with an atomic number greater than 137 would require its 1s electrons to be traveling faster than the speed of light. Even in the Dirac equation, which accounts for relativistic effects, the wave function of the electron for atoms with is oscillatory and unbounded. The significance of element 137, also known as untriseptium, was first pointed out by the physicist Richard Feynman. Element 137 is sometimes informally called feynmanium (symbol Fy).[37] However, Feynman's approximation fails to predict the exact critical value of Z due to the non-point-charge nature of the nucleus and very small orbital radius of inner electrons, resulting in a potential seen by inner electrons which is effectively less than Z. The critical Z value, which makes the atom unstable with regard to high-field breakdown of the vacuum and production of electron-positron pairs, does not occur until Z is about 173. These conditions are not seen except transiently in collisions of very heavy nuclei such as lead or uranium in accelerators, where such electron-positron production from these effects has been claimed to be observed.



{kind=link}
There are no nodes in relativistic orbital densities, although individual components of the wave function will have nodes.[38]
pp hybridization (conjectured)
In late period 8 elements, a hybrid of 8p3/2 and 9p1/2 is expected to exist,[39] where "3/2" and "1/2" refer to the total angular momentum quantum number. This "pp" hybrid may be responsible for the p-block of the period due to properties similar to p subshells in ordinary valence shells. Energy levels of 8p3/2 and 9p1/2 come close due to relativistic spin–orbit effects; the 9s subshell should also participate, as these elements are expected to be analogous to the respective 5p elements indium through xenon.
Transitions between orbitals
Bound quantum states have discrete energy levels. When applied to atomic orbitals, this means that the energy differences between states are also discrete. A transition between these states (i.e., an electron absorbing or emitting a photon) can thus happen only if the photon has an energy corresponding with the exact energy difference between said states.
Consider two states of the hydrogen atom:
- State n = 1, ℓ = 0, mℓ = 0 and ms = +1/2
- State n = 2, ℓ = 0, mℓ = 0 and ms = −1/2
By quantum theory, state 1 has a fixed energy of E1, and state 2 has a fixed energy of E2. Now, what would happen if an electron in state 1 were to move to state 2? For this to happen, the electron would need to gain an energy of exactly E2 − E1. If the electron receives energy that is less than or greater than this value, it cannot jump from state 1 to state 2. Now, suppose we irradiate the atom with a broad-spectrum of light. Photons that reach the atom that have an energy of exactly E2 − E1 will be absorbed by the electron in state 1, and that electron will jump to state 2. However, photons that are greater or lower in energy cannot be absorbed by the electron, because the electron can jump only to one of the orbitals, it cannot jump to a state between orbitals. The result is that only photons of a specific frequency will be absorbed by the atom. This creates a line in the spectrum, known as an absorption line, which corresponds to the energy difference between states 1 and 2.
The atomic orbital model thus predicts line spectra, which are observed experimentally. This is one of the main validations of the atomic orbital model.
The atomic orbital model is nevertheless an approximation to the full quantum theory, which only recognizes many electron states. The predictions of line spectra are qualitatively useful but are not quantitatively accurate for atoms and ions other than those containing only one electron.
See also
References
- ^ Orchin, Milton; Macomber, Roger S.; Pinhas, Allan; Wilson, R. Marshall (2005). "1. Atomic Orbital Theory" (PDF). The Vocabulary and Concepts of Organic Chemistry (2nd ed.). Wiley. Archived (PDF) from the original on 9 October 2022.
- ^ Daintith, J. (2004). Oxford Dictionary of Chemistry. New York: Oxford University Press. pp. 407–409. ISBN 978-0-19-860918-6.
- ^ Griffiths, David (1995). Introduction to Quantum Mechanics. Prentice Hall. pp. 190–191. ISBN 978-0-13-124405-4.
- ^ Levine, Ira (2000). Quantum Chemistry (5 ed.). Prentice Hall. pp. 144–145. ISBN 978-0-13-685512-5.
- ^ Laidler, Keith J.; Meiser, John H. (1982). Physical Chemistry. Benjamin/Cummings. p. 488. ISBN 978-0-8053-5682-3.
- ^ Atkins, Peter; de Paula, Julio; Friedman, Ronald (2009). Quanta, Matter, and Change: A Molecular Approach to Physical Chemistry. Oxford University Press. p. 106. ISBN 978-0-19-920606-3.
- ^ Feynman, Richard; Leighton, Robert B.; Sands, Matthew (2006). The Feynman Lectures on Physics – The Definitive Edition, Vol 1 lect 6. Pearson PLC, Addison Wesley. p. 11. ISBN 978-0-8053-9046-9.
- ^ Roger Penrose, The Road to Reality
- ^ Levine, Ira N. (1991). Quantum Chemistry (4th ed.). Prentice-Hall. p. 262. ISBN 0-205-12770-3.
Therefore, the wave function of a system of identical interacting particles must not distinguish among the particles.
- ^ Mulliken, Robert S. (July 1932). "Electronic Structures of Polyatomic Molecules and Valence. II. General Considerations". Physical Review. 41 (1): 49–71. Bibcode:1932PhRv...41...49M. doi:10.1103/PhysRev.41.49.
- ^ a b Bohr, Niels (1913). "On the Constitution of Atoms and Molecules". Philosophical Magazine. 26 (1): 476. Bibcode:1914Natur..93..268N. doi:10.1038/093268a0. S2CID 3977652.
- ^ a b Nagaoka, Hantaro (May 1904). "Kinetics of a System of Particles illustrating the Line and the Band Spectrum and the Phenomena of Radioactivity". Philosophical Magazine. 7 (41): 445–455. doi:10.1080/14786440409463141. Archived from the original on 27 November 2017. Retrieved 30 May 2009.
- ^ Bryson, Bill (2003). A Short History of Nearly Everything. Broadway Books. pp. 141–143. ISBN 978-0-7679-0818-4.
- ^ Thomson, J. J. (1897). "Cathode rays". Philosophical Magazine. 44 (269): 293. doi:10.1080/14786449708621070.
- ^ Thomson, J. J. (1904). "On the Structure of the Atom: an Investigation of the Stability and Periods of Oscillation of a number of Corpuscles arranged at equal intervals around the Circumference of a Circle; with Application of the Results to the Theory of Atomic Structure" (extract of paper). Philosophical Magazine. Series 6. 7 (39): 237–265. doi:10.1080/14786440409463107.
- ^ Rhodes, Richard (1995). The Making of the Atomic Bomb. Simon & Schuster. pp. 50–51. ISBN 978-0-684-81378-3.
- ^ Nagaoka, Hantaro (May 1904). "Kinetics of a System of Particles illustrating the Line and the Band Spectrum and the Phenomena of Radioactivity". Philosophical Magazine. 7 (41): 446. doi:10.1080/14786440409463141. Archived from the original on 27 November 2017. Retrieved 30 May 2009.
- ^ Heisenberg, W. (March 1927). "Über den anschaulichen Inhalt der quantentheoretischen Kinematik und Mechanik". Zeitschrift für Physik A. 43 (3–4): 172–198. Bibcode:1927ZPhy...43..172H. doi:10.1007/BF01397280. S2CID 122763326.
- ^ Bohr, Niels (April 1928). "The Quantum Postulate and the Recent Development of Atomic Theory". Nature. 121 (3050): 580–590. Bibcode:1928Natur.121..580B. doi:10.1038/121580a0.
- ^ Greiner, Walter (1994). Quantum mechanics : Introduction (2nd corrected ed.). New York, Berlin, Heidelberg: Springer-Verlag. p. 163. ISBN 978-0-387-58080-7.
- ^ Gerlach, W.; Stern, O. (1922). "Das magnetische Moment des Silberatoms". Zeitschrift für Physik. 9 (1): 353–355. Bibcode:1922ZPhy....9..353G. doi:10.1007/BF01326984. S2CID 126109346.
- ^ Thaller, Bernd (2004). Advanced visual quantum mechanics. New York: Springer/TELOS. ISBN 978-0387207773.
- ^ Petrucci, Ralph; Herring, F.; Madura, Jeffry; Bissonnette, Carey (2016). General chemistry: principles and modern applications (11th ed.). [Place of publication not identified]: Prentice Hall. ISBN 978-0133897319.
- ^ Messiah, Albert (1999). Quantum mechanics : two volumes bound as one (Two vol. bound as one, unabridged reprint ed.). Mineola, NY: Dover. ISBN 978-0-486-40924-5.
- ^ Claude Cohen-Tannoudji; Bernard Diu; Franck Laloë; et al. (1996). Quantum mechanics. Translated by from the French by Susan Reid Hemley. Wiley-Interscience. ISBN 978-0-471-56952-7.
- ^ Levine, Ira (2014). Quantum Chemistry (7th ed.). Pearson Education. pp. 141–2. ISBN 978-0-321-80345-0.
- ^ Blanco, Miguel A.; Flórez, M.; Bermejo, M. (December 1997). "Evaluation of the rotation matrices in the basis of real spherical harmonics". Journal of Molecular Structure: THEOCHEM. 419 (1–3): 19–27. doi:10.1016/S0166-1280(97)00185-1.
- ^ General chemistry : principles and modern applications. [Place of publication not identified]: Prentice Hall. 2016. ISBN 978-0133897319.
- ^ Friedman (1964). "The shapes of the f orbitals". J. Chem. Educ. 41 (7): 354. Bibcode:1964JChEd..41..354F. doi:10.1021/ed041p354.
- ^ Stanley Raimes (1963). The Wave Mechanics Of Electrons In Metals. North-Holland Publishing Company - Amsterdam. p. 39.
- ^ a b Jeong, Jong Seok; Odlyzko, Michael L.; Xu, Peng; Jalan, Bharat; Mkhoyan, K. Andre (26 April 2016). "Probing core-electron orbitals by scanning transmission electron microscopy and measuring the delocalization of core-level excitations". Physical Review B. 93 (16): 165140. Bibcode:2016PhRvB..93p5140J. doi:10.1103/PhysRevB.93.165140.
- ^ Powell, Richard E. (1968). "The five equivalent d orbitals". Journal of Chemical Education. 45 (1): 45. Bibcode:1968JChEd..45...45P. doi:10.1021/ed045p45.
- ^ Kimball, George E. (1940). "Directed Valence". The Journal of Chemical Physics. 8 (2): 188. Bibcode:1940JChPh...8..188K. doi:10.1063/1.1750628.
- ^ Cazenave, Lions, T., P.; Lions, P. L. (1982). "Orbital stability of standing waves for some nonlinear Schrödinger equations". Communications in Mathematical Physics. 85 (4): 549–561. Bibcode:1982CMaPh..85..549C. doi:10.1007/BF01403504. S2CID 120472894.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Bohr, Niels (1923). "Über die Anwendung der Quantumtheorie auf den Atombau. I". Zeitschrift für Physik. 13 (1): 117. Bibcode:1923ZPhy...13..117B. doi:10.1007/BF01328209. S2CID 123582460.
- ^ Lower, Stephen. "Primer on Quantum Theory of the Atom".
- ^ Poliakoff, Martyn; Tang, Samantha (9 February 2015). "The periodic table: icon and inspiration". Philosophical Transactions of the Royal Society A. 373 (2037): 20140211. Bibcode:2015RSPTA.37340211P. doi:10.1098/rsta.2014.0211. PMID 25666072.
- ^ Szabo, Attila (1969). "Contour diagrams for relativistic orbitals". Journal of Chemical Education. 46 (10): 678. Bibcode:1969JChEd..46..678S. doi:10.1021/ed046p678.
- ^ Fricke, Burkhard (1975). Superheavy elements: a prediction of their chemical and physical properties. Structure and Bonding. Vol. 21. pp. 89–144. doi:10.1007/BFb0116498. ISBN 978-3-540-07109-9. Retrieved 4 October 2013.
{{cite book}}:|journal=ignored (help)
- McCaw, Charles S. (2015). Orbitals: With Applications in Atomic Spectra. Singapore: World Scientific Publishing Company. ISBN 9781783264162.
- Tipler, Paul; Llewellyn, Ralph (2003). Modern Physics (4 ed.). New York: W. H. Freeman and Company. ISBN 978-0-7167-4345-3.
- Scerri, Eric (2007). The Periodic Table, Its Story and Its Significance. New York: Oxford University Press. ISBN 978-0-19-530573-9.
- Levine, Ira (2014). Quantum Chemistry (7th ed.). Pearson Education. ISBN 978-0-321-80345-0.
- Griffiths, David (2000). Introduction to Quantum Mechanics (2 ed.). Benjamin Cummings. ISBN 978-0-13-111892-8.
- Cohen, Irwin; Bustard, Thomas (1966). "Atomic Orbitals: Limitations and Variations". J. Chem. Educ. 43 (4): 187. Bibcode:1966JChEd..43..187C. doi:10.1021/ed043p187.
External links
- 3D representation of hydrogenic orbitals
- The Orbitron, a visualization of all common and uncommon atomic orbitals, from 1s to 7g
- Grand table Still images of many orbitals
| Historic models |
|
|---|---|
| Current models |
|