

| |
 Co2(CO)8 soaked in hexanes
| |
| Names | |
|---|---|
| IUPAC name
Octacarbonyldicobalt(Co—Co)
| |
| Other names
Cobalt carbonyl (2:8), di-mu-Carbonylhexacarbonyldicobalt, Cobalt octacarbonyl, Cobalt tetracarbonyl dimer, Dicobalt carbonyl, Octacarbonyldicobalt
| |
| Identifiers | |
3D model (JSmol)
|
|
| ChemSpider | |
| ECHA InfoCard | 100.030.454 |
| EC Number |
|
PubChem CID
|
|
| RTECS number |
|
| UNII | |
| UN number | 3281 |
CompTox Dashboard (EPA)
|
|
| |
| |
| Properties | |
| Co2(CO)8 | |
| Molar mass | 341.95 g/mol |
| Appearance | red-orange crystals |
| Density | 1.87 g/cm3 |
| Melting point | 51 to 52 °C (124 to 126 °F; 324 to 325 K) |
| Boiling point | 52 °C (126 °F; 325 K) decomposes |
| insoluble | |
| Vapor pressure | 0.7 mmHg (20 °C)[1] |
| Structure | |
| 1.33 D (C2v isomer) 0 D (D3d isomer) | |
| Hazards | |
| Occupational safety and health (OHS/OSH): | |
Main hazards
|
Potential carcinogen |
| GHS labelling: | |
   
| |
| Danger | |
| H251, H302, H304, H315, H317, H330, H351, H361, H412 | |
| P201, P260, P273, P280, P304+P340+P310, P403+P233 | |
| NFPA 704 (fire diamond) | |
| Flash point | -23 °C (-9.4 °F)[1] |
| Lethal dose or concentration (LD, LC): | |
LD50 (median dose)
|
15 mg/kg (oral, rat) |
| NIOSH (US health exposure limits): | |
PEL (Permissible)
|
none[1] |
REL (Recommended)
|
TWA 0.1 mg/m3[1] |
IDLH (Immediate danger)
|
N.D.[1] |
| Safety data sheet (SDS) | External SDS |
| Related compounds | |
Related metal carbonyls
|
Iron pentacarbonyl Diiron nonacarbonyl Nickel tetracarbonyl |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
| |

Dicobalt octacarbonyl is an organocobalt compound with composition Co2(CO)8. This metal carbonyl is used as a reagent and catalyst in organometallic chemistry and organic synthesis, and is central to much known organocobalt chemistry.[2][3] It is the parent member of a family of hydroformylation catalysts.[4] Each molecule consists of two cobalt atoms bound to eight carbon monoxide ligands, although multiple structural isomers are known.[5] Some of the carbonyl ligands are labile.
YouTube Encyclopedic
-
1/3Views:20 166314780
-
Mod-02 Lec-02 Metal carbonyl complexes
-
L-04- Oxo process or Hydroformylation Reaction( Preparation of aldehyde from alkenes)
-
Mod-06 Lec-28 Reactivity changes in coordinated ligands
Transcription
Today, we will discuss one of the most common ligands in organometallic chemistry and that is carbon monoxide. Carbon monoxide is probably most frequently encountered in organometallic chemistry and is significant portion of literature that is available today in organometallic chemistry, deals with at least 1 carbon monoxide ligand in the coordination sphere of the metal. So, when we look at carbon monoxide and the type of complexes it forms, it is interesting to that almost the whole periodic table can be involved in this chemistry of carbon monoxide. And metals significantly we are going to talk about transition metal carbon monoxide complexes. Another point of interest is the fact, that all the transition metals and when we say transition metals in this color coded periodic table, that we see here the transition metals are the once that range from scandium to zinc. Specifically, if you look at the complexes that are formed, you can notice that they are all homoleptic complexes that can be generated using carbon monoxide. This is probably unique to carbon monoxide, not all the other ligands that we encounter in organometallic chemistry can form homoleptic complexes. By homoleptic complexes we mean those metal complexes, in which only 1 ligand is involved in the coordination sphere of the metal. So, if you look at the first row of the representative row of the transition metals, so that goes from scandium to zinc. And we can, we will consider only the elements that are forming good carbon monoxide complexes, and those are in the range from vanadium to copper. So, from vanadium to copper you do have frequent occurrence of carbon monoxide complexes. We will see in a moment why this is the case; all of them are capable of forming homoleptic or M C O n complexes. So, these are complexes, which have M C O n and only 1 ligand is formed in the coordination sphere of the metal. Let us just take a look at the systems that we have here on this table, you will notice that you have for all the metals, which have even number of electrons that is chromium, iron and nickel, you have no charges on them. In other words since, carbon monoxide is a neutral ligand and you have n number of carbon monoxides it does not add any charge to the metal the metal is found in the 0 oxidation state. So, metal is in the 0 oxidation state and you have n number of carbon monoxides around them. And so, if you take the metals which are there with odd number of electrons and so that this vanadium has got 5 d electrons. And similarly, manganese has got 7 valence electrons in the d shell, so that might be d 5 s 2 or you might consider it as a total of 7 electrons in the valence shell and cobalt accordingly will have 9 valence electrons. So, the systems which have odd number of electrons tend to form complexes, which have a charge or if you go to classical coordination chemistry you will notice that the metals are usually found in positive oxidation states. In these systems, where we have these odd number of electrons surprisingly you have a tendency to form negatively charged complexes. In other words the net charge on the complex is minus 1 in many of these cases and if it is minus 1 then since, carbon monoxide is neutral the metal should be in a negative oxidation state. This is a rather strange instance, because metal is usually considered an electropositive element. So, one would not expect it to form a negatively charged species, so this is one of the first factors that we have to take into consideration, when we look at the chemistry of transition metal carbon carbonyls. So, except there is one exception here, which is the copper and we will we will try to explain that as time goes on. So, metal is in the 0 oxidation state and we also notice that the coordination number and the geometry appear to be restricted. Let us take a look at the coordination number you will notice that in coordination chemistry Werner coordination, Werner’s chemistry you will notice that the metal is in a positive coordination state. And you will also notice that many of the complexes are either octahedral, or tetrahedral not many other geometries are encountered in coordination chemistry. Unlike that, in organometallic chemistry you seem to be having a restriction on the coordination number and the geometry So, the next aspect that we have to consider is the oxidation state and the coordination. So, this is something which we have just mentioned the metal is found in the 0 oxidation state and the coordination number and the geometry are well restricted. Specifically, if you look at the compounds that are there in the first row, if you look only at the mononuclear complexes they all have a variable geometry. Nevertheless, it is distinctly attached to the number of carbon monoxide ligands that are present. In other words, if I have 5 carbon monoxides present in the coordination sphere of the metal, then the 5 carbon monoxides are distributed in a symmetrical fashion around the metal. If they, if I have 4 carbon monoxides distributed around the metal, then the 4 carbon monoxides are in a tetrahedral environment around the metal. Surprisingly, when you talk about these negatively charged systems they are also capable of forming dimeric complexes. Here are 2 instances, where you have 2 dimeric complexes that are formed, it looks as if you can have some other ways of compensating for the number of electrons around the metal. If indeed the metal was forming this cobalt tetracobaltate anion, to compensate for the number of electrons it was having in the valence shell. Then there seems to be another way of forming the same number of valence electrons in the complex and that seems to be in the form of making a dimeric species. Interestingly, there is one species in this whole series which is iron which forms a dimer. Nevertheless, there is a way of making the neutral complex with the right number of carbon monoxides around it. So, even considering the fact that you have a series of complexes right from vanadium to copper, which are mononuclear or charged. You find that the dimeric species are mostly in the neutral state, but they can also be generated by reacting them with a very good electron source like sodium amalgam or potassium amalgam. You can inject electrons into these dimeric species and also generate negatively charged. So, negatively charged metal complexes are formed in organometallic chemistry, which is very different from Werner’s coordination chemistry where it is very rare. Now, what we have noticed in the series of complexes that we have just encountered is that all of them end up with a total of 18 electrons around the metal atom. This was the reason why people started talking about the 18 electron rule. Now, the 18 electrons were generated by adding the number of electrons around the metal to the total number of electrons donated by the ligands. In Werner chemistry, in Werner’s coordination chemistry, what you find is that you do not have this strict following of 18 electrons around the valence shell. Here for example, I have noted that cobalt 3 plus, cobalt 3 plus has a valence number electron number of valence electrons equal to 6. So, that has got 6 valence electrons it is a d 6 complex. Now, if it combines with a water molecule 6 water molecules then you can form a very stable coordination compound, which is cobalt hexa aqua cobalt. Cobalt 3 complex and that is denoted here. Now, you will notice that total number of valence electrons around the cobalt also equal 18, 6 electrons from cobalt and 6 into 2, 12 electrons from the water molecules, so a total of 18 electrons can be generated a total of 18 electrons can be generated on the cobalt ion. Unlike, the cobalt hexa eco complex a very similar complex with chromium 3 plus now has got only d 3 valence electrons, so it has got 3 valence electrons. So, 3 valence electrons on the chromium plus 6 into 2, 12 electrons from the water molecule. So, you have a total of 15 valence electrons on this complex. So, you can see that Werner complex is, even if they form very stable systems they like to be octahedral or tetrahedral, but they do not follow the 18 electron rule or they do not seem to be having an 18 electron rule governing their electronic structure. Unlike this, the coordination unlike the coordination compounds formed in Werner chemistry organometallic systems tend to form 18 electron complexes. Let us just go back and look at the complexes that we have generated especially the neutral ones. It is very easy to do the electron counting in these cases. Chromium for example, we have just noted and has got a total of 6 valence electrons, carbon monoxide gives 2 electrons, so 6 into 2 will also give you 12 electrons. So, 6 plus 12 gives you 18 valence electrons a total of 18 valence electrons are there if you take iron-iron will give you eight valence electrons. And carbon monoxide 5 carbon monoxides can give you a total of 10 electrons. So, again a total of 18 electrons are generated around the iron. If you count the valence electrons which are available for each of these metal atoms you will always end up with a total of 18 electrons. This is true of nickel, it is true of iron, chromium and even the vanadium case, where you have 5 valence electrons on the vanadium and so you have to add an extra electron to the vanadium in order to generate the carbonyl complex. In the case of copper, you have a positive charge and this is in fact a unique system, it does not seem to follow the 18 valence electron rule, but you can generate a tetracoordinate C u C O 4 complex, also and that is possible. And people have generated a range of complexes in this case, but the number of complexes which are positively charged, and have only carbon monoxide in the coordination sphere are rather small. Copper, gold, silver are some of the species which form positively charged uh complexes and you will notice that if it is Cu C O 4, then it should have a valence electron count of 18 also. So, let us proceed further what we have noted is that the organometallic complexes that we are encountering in the systems strictly follow an 18 electron rule. And this is very different from the Werner’s coordination chemistry, where you only have an octahedral complex being formed or a tetrahedral complex. The driving force seems to be the restriction on the coordination number, and geometry rather than on the valence electron count. Now, you will notice that in the complexes the Werner’s coordination compounds that we have encountered you have a variety of oxidation states. You might have a nickel 2 plus, H 2 O 6, you can have a cobalt 2 plus, H 2 O 6. So, it can be a cobalt 2 plus aqua complex or it can be a nickel 2 plus aqua complex and these complexes can be nicely colored. Here, you can see them with very bright colors blue, green and pink and these complexes tend to be very highly colored. On the other hand, if you take chromium carbonyl complex, which we just talked about in C r C O 6, this is neutral and it is also interesting to note that it is a colorless species. So, although the oxidation states is one of the differences it is also interesting that the color of these species tend to be completely colorless, or at the most slightly colored at yellow in the case of iron carbonyl like Fe CO 5, is light yellow in color. So, these complexes have a different color and they also have different oxidation states compared to classical coordination chemistry, so nickel tends to form only 2 plus complexes, but cobalt can be cobalt 2 plus it can also be cobalt 3 plus. So, variable oxidation state is the characteristic of Werner’s chemistry this is typical of Werner’s chemistry. Whereas, organometallic chemistry tends to stick with simple neutral complexes when it comes to carbonyl, homoleptic carbonyl complexes. This is also to be noted the fact that magnetic properties of the Werner complexes are often either diamagnetic or paramagnetic. They tend to vary it tends depends on the oxidation state it tends to depend on the strength of the ligand. If you have very strong ligands, they tend to form low spin complexes or even diamagnetic complexes if the electron count is right. Whereas, in the case of organometallic complexes almost all of them tend to be diamagnetic. This is a distinct advantage, because you can carry out N M R spectroscopy of these complexes. N M R spectroscopy allows you to monitor these compounds in situ in a very easy fashion and this advantage is not present for the coordination chemistry, the Werner coordination chemistry that we encounter. When you have paramagnetic systems and paramagnetic systems tend to destroy the N M R signals fairly easily. Vanadium carbonyl is an exception, because that V C O 6 can be generated and if it is V C O 6 it tends to be paramagnetic it is not diamagnetic, so this is one exception that we note, okay? So, let us move on I mentioned earlier that metal carbonyls tend to form reasonably symmetric structures. They are not necessarily tetrahedral, octahedral although these tetrahedral and octahedral complexes are indeed observed typical examples are nickel tetracarbonyl, which is probably the one that chemists study encounter first, because that is very easy to synthesize and chromium hexacarbonyl. So, that is another molecule and that has got a nice octahedral complex, it is a nice octahedral geometry. But, you will notice that the complexes are all having carbon monoxides symmetrically distributed around the metal this seems to be a hallmark of the type of compounds that are formed by metal carbonyls. So, apart from these mononuclear complexes it is also possible to form multinuclear complexes with metal-metal bonds. Metal-metal bonds are reasonably rare in coordination chemistry in the case of Werner’s chemistry you normally do not encounter metal-metal bonds. And if you do, if you do you do have the possibility of a bridging ligand in addition to the metal bond, but in the case of carbonyl chemistry. In the case of organometallic compounds, which are supported by carbon monoxide you see that metal-metal bonds are reasonably common. And a variety of complexes can be generated which have got both a metal-metal bond and also a bridging carbon monoxide. First, let us take a look at those complexes that have a metal-metal bond. So, here is a set of complexes that I have pictured here and these compounds have got they strictly follow the 18 electron rule, still follow the 18 electron rule. They have carbon monoxides, which are just indicated by a straight line here. So, for example, so for example, if you have if you choose this complex each one of these lines, which I have, dawn here represents a carbon monoxide, in order to make the structure readily understandable. All the carbon monoxides are just shown as sticks and so each one of these lines represent each one of these lines represent a carbon monoxide. So, each metal centre has got 4 carbon monoxides, each metal centre has got 4 carbon monoxides in this case and so you form M 3 C O 12 complex and this is very common for all the transition metals. In the case of ruthenium and osmium you can form a system where you have a metal-metal bond supporting the formation of a trimeric species. Here you have another instance of a tetrameric species and this is a system where you have iridium with 3 carbon monoxides. Each of these vertices of these tetrahedra of these tetrahedran, this tetrahedran that you have here each one of these vertices is capped by an I r C O 3 moiety so each one of these vertices is occupied by an I r C O 3 moiety and that is the structure, which is shown here. In order to simplify the structure, we have shown it with only simple lines, another simple complex which is also having a metal-metal bond with no bridges. And this is unique for organometallic chemistry as I mentioned before, this is manganese decacarbonyl complex dimanganese decacarbonyl complex which is pictured here and this has got a single metal-metal bond which is holding the 2 units together. Once again you will notice that, because manganese has got 7 valence electrons manganese has 7 valence electrons you will need it is one electron short. If I form a complex like M n CO 5, if I form a complex like Mn C O 5 it is 1 electron short it is a 17 valence electron species. Now, these 17 valence electrons this one electron if it can be shared between the 2 manganese atoms then each manganese can end up with a valence electron count of 18. So, that sharing of that electron between the 2 manganese atoms results in the formation of a bond between the 2 manganese atoms. And that is what you have in each one of these cases either it forms 2 bonds as in the case of ruthenium, it has to form 2 bonds in order to achieve a valence electron count of 18. And in the case of iridium, it has to form 3 bonds in order to achieve a valence electron count of 18. So, the formation of a poly nuclear species with a metal-metal bond is primarily determined by the electron count this again brings us back to what we discussed earlier. And that is a fact these metal complexes are significantly driven by the tendency to form 18 electron species. The total valence electron has to be 18. So, another aspect I talked about when I mentioned poly nuclear systems is the fact that you can have bridging carbon monoxides. The fact that you can have bridging carbon monoxides is in again a unique factor, which is not found in coordination chemistry in the classical coordination chemistry. Carbon monoxide is a nice terminal ligand or in other words it can form it can donate 2 electrons and form a metal carbonyl bond and that is a terminal ligand. Surprisingly, carbon monoxide is found to be bridging in some of the dimers and trimers that can be generated. Especially in the case of the first row of transition metals. This is very different from water and ammonia water and ammonia are also, 2 electron donors N H 3 can donate 2 electrons water can also donate 2 electrons, but they do not form dimers like the metal carbonyls. Here, I have pictured a series of trimeric and tetrameric species and in each one of these systems you will encounter a bridging carbon monoxide. In some cases it is bridging 2 metal atoms in this case. For example, it is bridging 2 metal atoms and so we have pictured it as the vertex of, we have pictured it like this where the 2 lines meet that represents bridging carbon monoxide. In other systems where 3 lines meet you have a triple e bridging carbon monoxide. So, you have a triple e bridging carbon monoxide which is capping one of the phases of this octahedral species. So, you have 3 metal atoms, you have 3 metal atoms and a cap a trigonal phase is capped by a carbon monoxide unit. This, a triple e bridging carbon monoxide which you find in this particular unit right here. And you have a 2 metal atoms being bridged by 2 metal atom species, you will notice that in this particular instance we encountered another d 8 system forming a trimeric ruthenium. For example, R u 3 C O 12 was also there, but it did not have this bridging carbon monoxide. So, this has raised a lot of debate as to the facility with which carbon monoxide can bridge and why it bridges 3 d transition metals and not necessarily 4 d and 5 d elements. Both ruthenium and osmium do not form the bridge species, but instead we notice that they form only the trimeric species, where you have no bridges, but terminal carbon monoxides. So, here you have all terminal carbon monoxides and in the case of iron you tend to form 2 bridges. So iron has the same electron count, but you have 2 bridges and if you notice the number of carbon monoxides around the iron atom however is identical. So, the 18 electron count is always maintained 18 electron count around the metal is maintained, but the number of carbon monoxides can either be bridging or terminal. And you can still achieve the same electron count, because the bridging carbon monoxide donates 1 electron to the iron on one side, this counts for 1 electron and this counts for 1 electron this bond counts for 1 electron as well. So, the 2 electrons donated from carbon monoxide are now split between the 2 iron atoms. In the case of triple e bridging carbon monoxides electron counting is little more difficult. However, we say that the 2 electrons are shared between 3 metal centers, so we have to do a total valence electron count and then take care of the number of electrons available for the metal atom. So, aggregation in the formation of multinuclear complexes is very commonly encountered in organometallic chemistry and in all these cases, you can have bridging and terminal carbon monoxides in the carbon monoxide chemistry that we have just seen and in one unique instance at least. At least in the case of cobalt, tricobalt, octacarbonyl which I have pictured here it has been noticed, but that both terminal and bridging modes of carbon monoxides are encountered in the complex. In the case of the solid state structure of C O 2 C O 8, which I have mentioned here C O 2 C O 8 in the solid state has got 2 bridges in the solution state, it has been noticed that there are no bridges in the dimeric species that is thus formed. So, it appears as if there is a very delicate balance between carbon monoxide being a bridging ligand or carbon monoxide being a terminal ligand, both of the systems appear to be equally facile. Again we have already encountered this the fact that you can have iron tricarbonyl, which has got 2 bridges and tricarbonyl has got 2 bridges and ruthenium tricarbonyl which has got no bridges. So, the fact that you can form bridging carbon monoxide complexes and complexes, where there are no bridges appears to indicate that there is a very small energy difference between the 2 states between the bridge state and the non bridge state. So, here I have pictured the two structures of the dicobalt octacarbonyl complex. In the dicobalt octacarbonyl complex in solution, you find there are no bridges and in the case of the solid state structure you have a very clear formation of a you have a very clear indication of a bridge structure. In the case of dicobalt monocarbonyl that is Fe 2 C O 9 you find that the 3 triple e-bridging structures and that in that involve the necessity of a metal-metal bond. In order to satisfy the electron count, because if you have 6 electrons donated by the carbon monoxides and you have let us do this electron count. So, each iron has got 8 electrons in its valence shell 3 carbonyls the terminal carbonyls, which I will mark as T C O they will give you 6 electrons. As well and if I have this bridging carbon monoxide, if that gives you 3 electrons to each iron, so you have a total of 17 valence electrons, a total of 17 valence electrons. So, as I mentioned earlier since we are 1 electron short, we are 1 electron short of the magic electron count of 18. You tend to form a iron-iron bond which will involve sharing of 1 electron from each one of these iron atoms. So, that a total valence electron count of 18 can be achieved, so the iron-iron bond will contribute to 1 electron that gives you a total of 18 valence electrons for the Fe 2 C O 9. So, this is chemistry that is completely governed by this tendency to form 18 valence electrons around the metal atom. Now, that we have considered the electronic structure around the metal atom. Let us take a brief look at some of the properties of these molecules. Werner coordination chemistry or Werner’s chemistry typically involved many ionic compounds and these ionic compounds are solids and in most instances they are solid complexes, very difficult to evaporate. On the other hand the physical properties of organometallic carbon monoxide complexes are completely different many of them, or most of them are liquids or gases and even if they are solids they can be readily sublimed. So, their vapor pressure is quite high and that is as a matter of concern, when you talk about handling them you need to be extremely careful, because they are quite volatile. Now, interestingly many of them are all soluble in organic solvents and that led to the rapid development of organometallic chemistry, because it was possible to use organic ligands, organic solvent in order to do the chemistry of organometallic compounds. It was possible to separate them from salts and coordination compounds fairly readily, because one could just wash it away. The negatively charged the other important fact that we need that. We have already noted, is the fact that negatively charged metal ions can be stabilized by carbon monoxide very readily those compounds are indeed ionic they are charged. They are usually solids and they might be colored as well, because of charge transfer phenomena. Let us now move on to the structure of these compounds, you find that the structure of these compounds are those that are governed by this 18 electron rule, but more than that they are not single iron metal carbon bonds. If you take the typical example of Fe C O 5, you find that the iron carbon bond that you would expect on the basis of the covalent radius of iron and the covalent single bond radius, bond of carbon. If you add these 2 together you would get a bond distance of 2.2 Angstroms. So, 2.2 Angstroms is what you would expect on the basis of adding the 2 single bond radii of the 2 elements iron and carbon, but the F e C O 5 molecule turns out to be having a iron carbon bond distance 1.83 Angstroms in the equatorial position. So, here is the molecule, which has a pentagonal 5 ligands it is got a trigonal bipyramidal geometry. Each one of these lines represents a carbon monoxide, so you have a carbon monoxide in each one of these points which ends at the end of this line and so, this bond distance is what we are talking about and this bond distance turns out to be smaller than, what you expect to be. On the basis of the single bond railings, the axial bond surprisingly is 1.81 Angstroms and the equatorial bond is the one, which is 1.83 Angstroms. So, these distances are longer than the free carbon mono, the carbon monoxide distances are the ones that we are talking about here C O distance. This distance is longer than what is observed in free carbon monoxide by point naught 1 Angstroms. So, you have two things that we need to explain. First of all the fact that all of them have got this 18 electron structure. Secondly you have to explain the fact that you have shorter bonds than you expect for a single bond. A single metal carbon bond and the third point that we note is the fact that you have a carbon oxygen bond distance, which is slightly longer. So, you have a very small elongation of the carbon monoxide bond, and you have a shortening of the iron carbon bond. Before we proceed further and explain the electronic structure of these complexes, I want to briefly mention the spectroscopy that can be done with these compounds. These compounds have got very significant carbon monoxide stretches, which change when their complex that is free carbon monoxide stretches that change when they are complex to the metal the free carbon stretch. Which is something that every organometallic student or every student of organometallic chemistry memorizes is the fact that it is 2143 centimeter minus 1. So, this magic number that you have with carbon monoxide is, because of the triple bond that you have between carbon and oxygen, so it is quite difficult to stretch this carbon oxygen bond. This carbon oxygen bond which is difficult to stretch appears at 2143 centimeter minus 1, but when it is complex to the metal it becomes easier to stretch this carbon and oxygen. This carbon oxygen stretching frequency drops down by 100 to almost 200 centimeters minus 1, when it is complex to the metal. This is another interesting phenomenon that we have to notice and that we have to explain. When we look at the electronic structure, but this gives us a very nice way of studying the structure of these molecules, because carbon monoxide stretch has got a very strong dipole moment change when it and so it can be readily observed in the infrared spectrum of these molecules. And we will look at some of the ways by which it can be used effectively. In a few compounds this carbon monoxide stretching frequency is not decreased, but it actually is increased and this is very rare, they this is quite rare this is centimeter minus 1 and those systems we find in the metal is positively charged. So, let us take a look at the bond dissociation energy of metal carbon carbonyl complexes. You will see here a series of numbers which are the, which are energy required to break the metal carbonyl bond chromium carbonyl. For example, requires an average of 25 kilocalories per mole to break this Cr C O bond. This bond is worth about 25 kilo calories per mole that is what we are saying in this table. So similarly, if you look at all the metals we find that the average bond dissociation energy is not varying much as you go from left to right in the periodic table. All of them have an average of approximately 25 to 26 calories kilo per mole. On the other hand as you go down the periodic table as you and go down the periodic table in the chromium series. For example, if you go from chromium hexacarbonyl to molybdenum hexacarbonyl, the bond energy seems to increase significantly it is almost increased by ten kilo calories per mole and that is about 33 percent increase and by another 16 percent increase, when you go from molybdenum to tungsten. So, it is possible to have very strong bonds in the case of 4 d and 5 d elements this happens to be a general phenomenon in organometallic chemistry the metal carbon bond strength, that you observe in most cases increases as you down the group. For the same group, if you have a bond strength of 100 kilo calories per mole, as you go down the group you will increase it by 150 or to 200 as you go to the 5 d series. So, the metal carbon bond becomes stronger as you go down the group. Let us come to the synthesis of metal carbonyls a little bit of chemistry. Before we go back to the understanding of metal carbonyl chemistry in terms of electronic structure, if you take iron powder and treat it with carbon monoxide at 200 atmospheric pressure. A high temperature relatively high temperature 200 degree celsius, You tend to form the volatile metal carbonyl complex which is Fe C O 5. This turns out be the simplest way to make iron carbonyl and it is fairly inexpensive, because carbon monoxide is cheap and iron powder is cheap, and you can make iron carbonyl in significant amount iron carbonyl. However is not readily available these days, because it turns out that you can make very interesting metal coatings, which can make. For example, aero plane invisible to the rador by making a coating of iron which is generated from iron carbonyl, which makes difficult to make or purchase iron carbonyl in spite of the fact that it is very easy to make. So, let us move on here we have iridium complex being generated with carbon monoxide c o d which is pictured here, is which is indicated here as c o d is in fact cyclooctadiene which is normally 15 cyclooctadiene and 15 cyclooctadiene and l is the ligand trimethylphosphene, which is easy to understand. So, if iridium has got these 2 ligands cyclooctadiene is a very weak ligand. We will encounter later in this lecture series it can be replaced by carbon monoxide to form this familiar complex, which is a square planar complex with Cl and CO and the trans geometry and 2 l ligands which are P Me 3 coordinated in a like fashion. So, this is the complex which you have here this is the complex that we are talking about and this is very readily generated using carbon monoxide. Carbon monoxide and c o d ligand which is that cyclooctadiene ligand. So, in the previous 2 examples we have generated the nickel, generated the metal carbonyl complex starting directly from the metal atom in the 0 oxidation state, or in the plus 1 oxidation state as in the case of iridium. Here we have started with nickel in the plus 2 oxidation state and we are going to reduce it using a compound, which is inorganic reducing agent. We can also reduce it using carbon monoxide itself carbon monoxide is a reducing agent, because we can oxidize carbon monoxide using the oxygen atoms, which are available in this oxide material and convert it to carbon dioxide. So, carbon monoxide is in fact a reducing agent, you can use carbon monoxide as a reducing agent or you can use an inorganic reducing agent as S 2 O 4 2 minus. So, either way you can generate a metal complex fairly readily by taking a slightly oxidized system a 2 plus species in that particular case nickel or even in the case of rhenium. You have a plus 7-oxidation state and it is you can still reduce it using carbon monoxide. To generate a metal carbonyl complex here is one example where we have a carbonyl which is a 0 oxidation state, which is in the 0 oxidation state this particular complex needs a little bit of explanation. Here you have 2 carbon monoxides trans to each other and you have 2 nitrogens, which are coordinated in this fashion with 2 methyl groups on the nitrogen that is this ligand t m e d a. You have 4 carbon monoxides, 2 carbon monoxides trans to each other and 2 carbon monoxides cis to each other. Now, if you treat this metal complex with sodium, sodium as I told you earlier can pump in electrons. Especially if it is used as an amalgam sodium and mercury turns out to be a very good reducing agent and it can generate, what we can call here as a chromium 2 minus complex, because the chromium the negative oxidation state of chromium. Here is in this particular case it is 4 minus 4 electrons are being pumped in and you have chromium and 4 minus oxidation state. So, these are unique complexes where you have a high negative charge on the metal atom. This is usually generated with either sodium and mercury or sodium as a molten sodium, that you can generate by slightly warming the sodium metal. Now, let us get back to the fact that the carbon monoxide stretching frequency has reduced from 2143 centimeter minus 1 to 1850 to 2100 approximately 100 to 150 centimeter minus 1 decrease a 100 to 2 00 minus 1 decrease in the stretching frequency. So, this decrease has to be explained when we look at the bonding. Let us go out and look at the bonding that is involved and we also, note the fact that this carbon monoxide stretching can vary depending on the type of carbon monoxide that you have. If it is a terminal carbon monoxide then the stretching frequency decreases significantly, and if it is bridging carbon monoxide then it is more like a ketonic carbon monoxide. The carbon monoxide stretching frequency can be as low as 1600 centimeter minus 1, when it is triple e bridging. As in the case of the compounds that we encounter the iridium polynuclear systems, we had triple e bridging carbon monoxides. In those cases the carbon monoxide is in fact more like a ketone or a C double bond O, or a C plus 0 minus species O, you have very low carbon oxygen stretching frequencies. It is very useful to use carbon monoxide to identify the type of compounds that they form for example, I have pictured here a M C O 2 system. One of them is trans here is a trans compound and here is a cis compound and the 2 carbon monoxides can be readily distinguished, because in the infrared spectrum of this molecule. You will observe for the trans isomer only a very strong signal, a single strong stretching frequency which is coming from the asymmetric stretching of the 2 carbon monoxide units, which will couple to each other. Here you have a symmetric stretch, where 2 carbon monoxide units are pulled in a symmetric fashion both of them are either pulled apart or in the other case where you have an asymmetric stretch. This is the new asymmetric stretch, then you have pulling apart of the 2 carbon and oxygen bonds pulling apart of the carbon oxygen bonds and in the other carbon oxygen, you have a compression of the C and O. So, this leads to an asymmetric stretch which is observed as a very strong band in the infrared spectrum, but the symmetric stretch is hardly is a very weak signal or if it is not observed in some cases. On the other hand, if you have a cis complex then you tend to have a strong band for the symmetric stretch and a strong band for the asymmetric stretch. So, carbon monoxide infrared spectra very useful. You can also use it for triple e bridging systems, here I have shown the 2 possibilities when you have carbon monoxide, you can in an octahedral complex. If you have triple e ligated systems then you have triple e carbon monoxide ligated systems, you can have the meridional isomer or the facial isomer the meridional isomer, will have 3 bands as pictured here, three infrared stretching frequencies. Usually the 2 asymmetric stretches that can have for this complex are strong bands and the symmetric stretches weak. Whereas, in the case of the facial isomer, you can have depending on the symmetry, you can have a small symmetric stretch and a very strong asymmetric stretch. So, based on the elemental formula you would be able to distinguish the M C O 2 and M CO 3 systems and once you know how many carbon monoxides are there, you can figure out the geometry of the complex. Whether it is cis or trans whether it is facial or meridional depending on the number of carbon monoxide bands that you have. Carbon monoxide complexes can also be studied using carbon 13 NMR spectroscopy and the only difficulty in these cases is the fact that the natural abundance of carbon is only 1 percent. Since, carbon is not a very sensitive nucleus, you tend to have very weak signals for the carbon monoxide in M C O, M C O N complexes. So, what one usually does is you add some agents external agents into the sample, so that the relaxation is really fast and so you can have reasonable signals for the carbonyl carbon, that is the 13 C in the carbonyl carbon. Now, we have considered a lot of things, lot of aspects of metal carbonyl chemistry. We have studied that they tend to form 18 electron complexes, they tend to form specific structures which are even negatively charged and they are very different from Werner coordination compounds. And they can be studied by infrared spectra, which indicate significant decrease in the carbon monoxide stretching frequencies. Lastly, I also want to ask this question, why do they form at all? Because carbon monoxide is a very stable molecule it is very difficult to generate isolated metal atoms, so isolated metal atoms are formed in the gas phase. If you vaporize them, but this requires a very large amount of energy, very often 1,000 to 2,000 k. If you heat a metal atom to a very high temperature then you can vaporize the metal. So, why is it that you can readily form metal carbonyl complexes in the case of nickel? You can even pass carbon monoxide at room temperature and atmospheric pressure and you can form nickel tetra carbonyl. So, these two complexes are even more interesting, you can readily form metal carbonyls at room temperature with carbon monoxide iron will form Fe C O 5 very readily. This can be done even at room temperature, using carbon monoxide with very pure iron, which can be readily distilled. This is the, these are volatile molecules which are readily distilled. So, carbon monoxide is a stable molecule it has got a very small dipole moment and the carbon end is negative and the protonation of carbon monoxide leads very little energy, but it does it can protonation carbon monoxide, that being the case why is that they form such stable compounds. In fact theory estimates that the bond dissociation energy of metal carbonyls can be as high as 70 to 100 kilo calories per mole. The carbon oxygen bond and the metal carbon bond are significantly different from what you would expect. So, all these things have to be explained. Let us take a look again at the structural parameters, we have already mentioned this that based on the iron carbon bond distance you expect a distance of 2.10. What you get is a much shorter bond distance and the carbon oxygen distance is in fact elongated. It is usually between 1.14 and 1.16 and you have a spectroscopic feature, which suggests that this frequency decreases. So, all these things have to be explained. One way to do this is to use molecular orbital theory, and we will use molecular orbital theory to explain the bonding in carbon monoxide in detail in the next lecture. But we will introduce this structure of carbon monoxide itself right now, and we will take a look at some of the valence orbitals. The valence orbitals of carbon monoxide are the ones, which have got a very large contribution from the carbon end. So, this is a carbon end and this is the oxygen end, and the valence orbital has got, these are only the sigma orbitals. And you can notice that the sigma orbital has got a very large contribution on the carbon end. These are the pi orbitals now, you have a pi and a pi star and this pi star also has large contribution on the carbon end. So, this is the introduction to the molecular orbitals of carbon monoxide. Now, we can see how these valence orbitals can interact with the metal in the next lecture. So, let us just summarize some of the aspects that we have learnt in metal carbonyl chemistry. So, these are the key features that we have encountered in today’s lecture. First of all, 18 electron complexes appear to dominate. There are very few complexes, in which you have very few complexes, in which you have more than or less than 18 electrons. You have also encountered the fact that neutral metal atoms, neutral metal atoms and not ions, as in Werner complexes are encountered. In Werner complexes, mostly they are charged systems. Thirdly carbon monoxide can bridge and form both terminal and bridging carbon monoxide units, and also we encountered several systems where there are metal-metal bonds, even when there are no bridging ligands. This is again uncommon in Werner’s chemistry. And we will note, we will study in the next lecture that the best explanation for all these factors is to use, what is called the DCD model, where there is a give and take of electrons. This appears to be a general phenomenon, and not just in metal carbonyl chemistry, but in the whole of organometallic chemistry.
Synthesis, structure, properties
Dicobalt octacarbonyl an orange-colored, pyrophoric solid.[6] It is synthesised by the high pressure carbonylation of cobalt(II) salts:[6]
- 2 (CH3COO)2Co + 8 CO + 2 H2 → Co2(CO)8 + 4 CH3COOH
The preparation is often carried out in the presence of cyanide, converting the cobalt(II) salt into a pentacyanocobaltate(II) complex that reacts with carbon monoxide to yield K[Co(CO)4]. Acidification produces cobalt tetracarbonyl hydride, HCo(CO)4, which degrades near room temperature to dicobalt octacarbonyl and hydrogen.[3][7] It can also be prepared by heating cobalt metal to above 250 °C in a stream of carbon monoxide gas at about 200 to 300 atm:[3]
- 2 Co + 8 CO → Co2(CO)8
It exist as a mixture of rapidly interconverting isomers.[2][3] In solution, there are two isomers known that rapidly interconvert:[5]

The major isomer (on the left in the above equilibrium process) contains two bridging carbonyl ligands linking the cobalt centres and six terminal carbonyl ligands, three on each metal.[5] It can be summarised by the formula (CO)3Co(μ-CO)2Co(CO)3 and has C2v symmetry. This structure resembles diiron nonacarbonyl (Fe2(CO)9) but with one fewer bridging carbonyl. The Co–Co distance is 2.52 Å, and the Co–COterminal and Co–CObridge distances are 1.80 and 1.90 Å, respectively.[8] Analysis of the bonding suggests the absence of a direct cobalt–cobalt bond.[9]
The minor isomer has no bridging carbonyl ligands, but instead has a direct bond between the cobalt centres and eight terminal carbonyl ligands, four on each metal atom.[5] It can be summarised by the formula (CO)4Co-Co(CO)4 and has D4d symmetry. It features an unbridged cobalt–cobalt bond that is 2.70 Å in length in the solid structure when crystallized together with C60.[10]
-
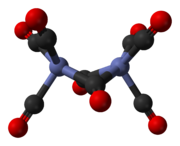 Bridged C2v isomer
Bridged C2v isomer -
 non-bridged D3d isomer
non-bridged D3d isomer -
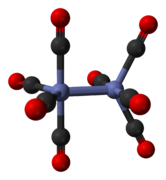 nonbridged D2d isomer
nonbridged D2d isomer



Reactions
Reduction
Dicobalt octacarbonyl is reductively cleaved by alkali metals and related reagents, such as sodium amalgam. The resulting salts protonate to give tetracarbonyl cobalt hydride:[3]
- Co2(CO)8 + 2 Na → 2 Na[Co(CO)4]
- Na[Co(CO)4] + H+ → H[Co(CO)4] + Na+
Salts of this form are also intermediates in the cyanide synthesis pathway for dicobalt octacarbonyl.[7]
Reactions with electrophiles
Halogens and related reagents cleave the Co–Co bond to give pentacoordinated halotetracarbonyls:
- Co2(CO)8 + Br2 → 2 Br[Co(CO)4]
Cobalt tricarbonyl nitrosyl is produced by treatment of dicobalt octacarbonyl with nitric oxide:
- Co2(CO)8 + 2 NO → 2 Co(CO)3NO + 2 CO
Reactions with alkynes
The Nicholas reaction is a substitution reaction whereby an alkoxy group located on the α-carbon of an alkyne is replaced by another nucleophile. The alkyne reacts first with dicobalt octacarbonyl, from which is generated a stabilized propargylic cation that reacts with the incoming nucleophile and the product then forms by oxidative demetallation.[11][12]

The Pauson–Khand reaction,[13] in which an alkyne, an alkene, and carbon monoxide cyclize to give a cyclopentenone, can be catalyzed by Co2(CO)8,[3][14] though newer methods that are more efficient have since been developed:[15][16]

Co2(CO)8 reacts with alkynes to form a stable covalent complex, which is useful as a protective group for the alkyne. This complex itself can also be used in the Pauson–Khand reaction.[13]
Intramolecular Pauson–Khand reactions, where the starting material contains both the alkene and alkyne moieties, are possible. In the asymmetric synthesis of the Lycopodium alkaloid huperzine-Q, Takayama and co-workers used an intramolecular Pauson–Khand reaction to cyclise an enyne containing a tert-butyldiphenylsilyl (TBDPS) protected primary alcohol.[17] The preparation of the cyclic siloxane moiety immediately prior to the introduction of the dicobalt octacarbonyl ensures that the product is formed with the desired conformation.[18]

Dicobalt octacarbonyl can catalyze alkyne trimerisation of diphenylacetylene and its derivatives to hexaphenylbenzenes.[19] Symmetrical diphenylacetylenes form 6-substituted hexaphenylbenzenes, while asymmetrical diphenylacetylenes form a mixture of two isomers.[20]


Hydroformylation

- 1
- Carbon monoxide dissociates from cobalt tetracarbonyl hydride to form the active catalyst, HCo(CO)3
- 2
- The cobalt centre π bonds to the alkene
- 3
- The alkene ligand inserts into the cobalt–hydride bond
- 4
- An additional carbonyl ligand coordinates
- 5
- A carbonyl ligand migrates into the cobalt–alkyl bond[21]
- 6
- Dihydrogen adds to the acyl complex
- 7
- The dihidrydo complex eliminates the aldehyde product,[22] regenerating the catalyst
- 8
- An unproductive and reversible side reaction
Hydrogenation of Co2(CO)8 produces cobalt tetracarbonyl hydride H[Co(CO)4]:[23]
- Co2(CO)8 + H2 → 2 H[Co(CO)4]
This hydride is a catalyst for hydroformylation – the conversion of alkenes to aldehydes.[4][23] The catalytic cycle for this hydroformylation is shown in the diagram.[4][21][22]
Substitution reactions
The CO ligands can be replaced with tertiary phosphine ligands to give Co2(CO)−8x(PR3)x. These bulky derivatives are more selective catalysts for hydroformylation reactions.[3] "Hard" Lewis bases, e.g. pyridine, cause disproportionation:
- 12 C5H5N + 3 Co2(CO)8 → 2 [Co(C5H5N)6][Co(CO)4]2 + 8 CO

Conversion to higher carbonyls
Heating causes decarbonylation and formation of tetracobalt dodecacarbonyl:[3][24]
- 2 Co2(CO)8 → Co4(CO)12 + 4 CO
Like many metal carbonyls, dicobalt octacarbonyl abstracts halides from alkyl halides. Upon reaction with bromoform, it converts to methylidynetricobaltnonacarbonyl, HCCo3(CO)9, by a reaction that can be idealised as:[25]
- 9 Co2(CO)8 + 4 CHBr3 → 4 HCCo3(CO)9 + 36 CO + 6 CoBr2
Safety
Co2(CO)8 a volatile source of cobalt(0), is pyrophoric and releases carbon monoxide upon decomposition.[26] The National Institute for Occupational Safety and Health has recommended that workers should not be exposed to concentrations greater than 0.1 mg/m3 over an eight-hour time-weighted average, without the proper respiratory gear.[27]
References
- ^ a b c d e NIOSH Pocket Guide to Chemical Hazards. "#0147". National Institute for Occupational Safety and Health (NIOSH).
- ^ a b Pauson, Peter L.; Stambuli, James P.; Chou, Teh-Chang; Hong, Bor-Cherng (2014). "Octacarbonyldicobalt". Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons. pp. 1–26. doi:10.1002/047084289X.ro001.pub3. ISBN 9780470842898.
- ^ a b c d e f g h Donaldson, John Dallas; Beyersmann, Detmar (2005). "Cobalt and Cobalt Compounds". Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH. doi:10.1002/14356007.a07_281.pub2. ISBN 3527306730.
- ^ a b c d Elschenbroich, C.; Salzer, A. (1992). Organometallics: A Concise Introduction (2nd ed.). Weinheim: Wiley-VCH. ISBN 3-527-28165-7.
- ^ a b c d Sweany, Ray L.; Brown, Theodore L. (1977). "Infrared spectra of matrix-isolated dicobalt octacarbonyl. Evidence for the third isomer". Inorganic Chemistry. 16 (2): 415–421. doi:10.1021/ic50168a037.
- ^ a b Gilmont, Paul; Blanchard, Arthur A. (1946). Dicobalt Octacarbonyl, Cobalt Nitrosyl Tricarbonyl, and Cobalt Tetracarbonyl Hydride. Inorganic Syntheses. Vol. 2. pp. 238–243. doi:10.1002/9780470132333.ch76. ISBN 9780470132333.
- ^ a b Orchin, Milton (1953). "Hydrogenation of Organic Compounds with Synthesis Gas". Advances in Catalysis. Vol. 5. Academic Press. pp. 385–415. ISBN 9780080565095.
- ^ Sumner, G. Gardner; Klug, Harold P.; Alexander, Leroy E. (1964). "The crystal structure of dicobalt octacarbonyl". Acta Crystallographica. 17 (6): 732–742. doi:10.1107/S0365110X64001803.
- ^ Green, Jennifer C.; Green, Malcolm L. H.; Parkin, Gerard (2012). "The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds". Chemical Communications. 2012 (94): 11481–11503. doi:10.1039/c2cc35304k. PMID 23047247.
- ^ Garcia, Thelma Y.; Fettinger, James C.; Olmstead, Marilyn M.; Balch, Alan L. (2009). "Splendid symmetry: Crystallization of an unbridged isomer of Co2(CO)8 in Co2(CO)8·C60". Chemical Communications. 2009 (46): 7143–7145. doi:10.1039/b915083h. PMID 19921010.
- ^ Nicholas, Kenneth M. (1987). "Chemistry and synthetic utility of cobalt-complexed propargyl cations". Acc. Chem. Res. (Review). 20 (6): 207–214. doi:10.1021/ar00138a001.
- ^ Teobald, Barry J. (2002). "The Nicholas reaction: The use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis". Tetrahedron (Review). 58 (21): 4133–4170. doi:10.1016/S0040-4020(02)00315-0.
- ^ a b Pauson, P. L.; Khand, I. U. (1977). "Uses of Cobalt-Carbonyl Acetylene Complexes in Organic Synthesis". Ann. N. Y. Acad. Sci. 295 (1): 2–14. Bibcode:1977NYASA.295....2P. doi:10.1111/j.1749-6632.1977.tb41819.x. S2CID 84203764.
- ^ Blanco-Urgoiti, Jaime; Añorbe, Loreto; Pérez-Serrano, Leticia; Domínguez, Gema; Pérez-Castells, Javier (2004). "The Pauson–Khand reaction, a powerful synthetic tool for the synthesis of complex molecules". Chem. Soc. Rev. 33 (1): 32–42. doi:10.1039/b300976a. PMID 14737507.
- ^ Schore, Neil E. (1991). "The Pauson–Khand Cycloaddition Reaction for Synthesis of Cyclopentenones". Org. React. 40: 1–90. doi:10.1002/0471264180.or040.01. ISBN 0471264180.
- ^ Gibson, Susan E.; Stevenazzi, Andrea (2003). "The Pauson–Khand Reaction: The Catalytic Age Is Here!". Angew. Chem. Int. Ed. 42 (16): 1800–1810. doi:10.1002/anie.200200547. PMID 12722067.
- ^ Nakayama, Atsushi; Kogure, Noriyuki; Kitajima, Mariko; Takayama, Hiromitsu (2011). "Asymmetric Total Synthesis of a Pentacyclic Lycopodium Alkaloid: Huperzine-Q". Angew. Chem. Int. Ed. 50 (35): 8025–8028. doi:10.1002/anie.201103550. PMID 21751323.
- ^ Ho, Tse-Lok (2016). "Dicobalt Octacarbonyl". Fiesers' Reagents for Organic Synthesis. Vol. 28. John Wiley & Sons. pp. 251–252. ISBN 9781118942819.
- ^ Vij, V.; Bhalla, V.; Kumar, M. (8 August 2016). "Hexaarylbenzene: Evolution of Properties and Applications of Multitalented Scaffold". Chemical Reviews. 116 (16): 9565–9627. doi:10.1021/acs.chemrev.6b00144.
- ^ Xiao, W.; Feng, X.; Ruffieux, P.; Gröning, O.; Müllen, K.; Fasel, R. (18 June 2008). "Self-Assembly of Chiral Molecular Honeycomb Networks on Au(111)". Journal of the American Chemical Society. 130 (28): 8910–8912. doi:10.1021/ja7106542.
- ^ a b Heck, Richard F.; Breslow, David S. (1961). "The Reaction of Cobalt Hydrotetracarbonyl with Olefins". Journal of the American Chemical Society. 83 (19): 4023–4027. doi:10.1021/ja01480a017.
- ^ a b Halpern, Jack (2001). "Organometallic chemistry at the threshold of a new millennium. Retrospect and prospect". Pure and Applied Chemistry. 73 (2): 209–220. doi:10.1351/pac200173020209.
- ^ a b Pfeffer, M.; Grellier, M. (2007). "Cobalt Organometallics". Comprehensive Organometallic Chemistry III. Vol. 7. Elsevier. pp. 1–119. doi:10.1016/B0-08-045047-4/00096-0. ISBN 9780080450476.
- ^ Chini, P. (1968). "The closed metal carbonyl clusters". Inorganica Chimica Acta Reviews. 2: 31–51. doi:10.1016/0073-8085(68)80013-0.
- ^ Nestle, Mara O.; Hallgren, John E.; Seyferth, Dietmar; Dawson, Peter; Robinson, Brian H. (2007). "μ3-Methylidyne and μ3-Benzylidyne-Tris(Tricarbonylcobalt)". Inorg. Synth. 20: 226–229. doi:10.1002/9780470132517.ch53. ISBN 9780470132517.
- ^ Cole Parmer MSDS
- ^ CDC - NIOSH Pocket Guide to Chemical Hazards